[](https://github.com/neherlab/treetime/actions/workflows/ci.yml)
[](https://anaconda.org/bioconda/treetime)
[](https://treetime.readthedocs.io/en/latest/)
## TreeTime: maximum likelihood dating and ancestral sequence inference
### Overview
TreeTime provides routines for ancestral sequence reconstruction and inference of molecular-clock phylogenies, i.e., a tree where all branches are scaled such that the positions of terminal nodes correspond to their sampling times and internal nodes are placed at the most likely time of divergence.
To optimize the likelihood of time-scaled phylogenies, TreeTime uses an iterative approach that first infers ancestral sequences given the branch length of the tree (assuming the branch length of the input tree is in units of average number of nucleotide or protein substitutions per site in the sequence). After infering ancestral sequences TreeTime optimizes the positions of unconstrained nodes on the time axis, and then repeats this cycle.
The only topology optimization are (optional) resolution of polytomies in a way that is most (approximately) consistent with the sampling time constraints on the tree.
The package is designed to be used as a stand-alone tool on the command-line or as a library used in larger phylogenetic analysis work-flows.
[The documentation of TreeTime is hosted on readthedocs.org](https://treetime.readthedocs.io/en/latest/).
In addition to scripting TreeTime or using it via the command-line, there is also a small web server at [treetime.ch](https://treetime.biozentrum.unibas.ch/).
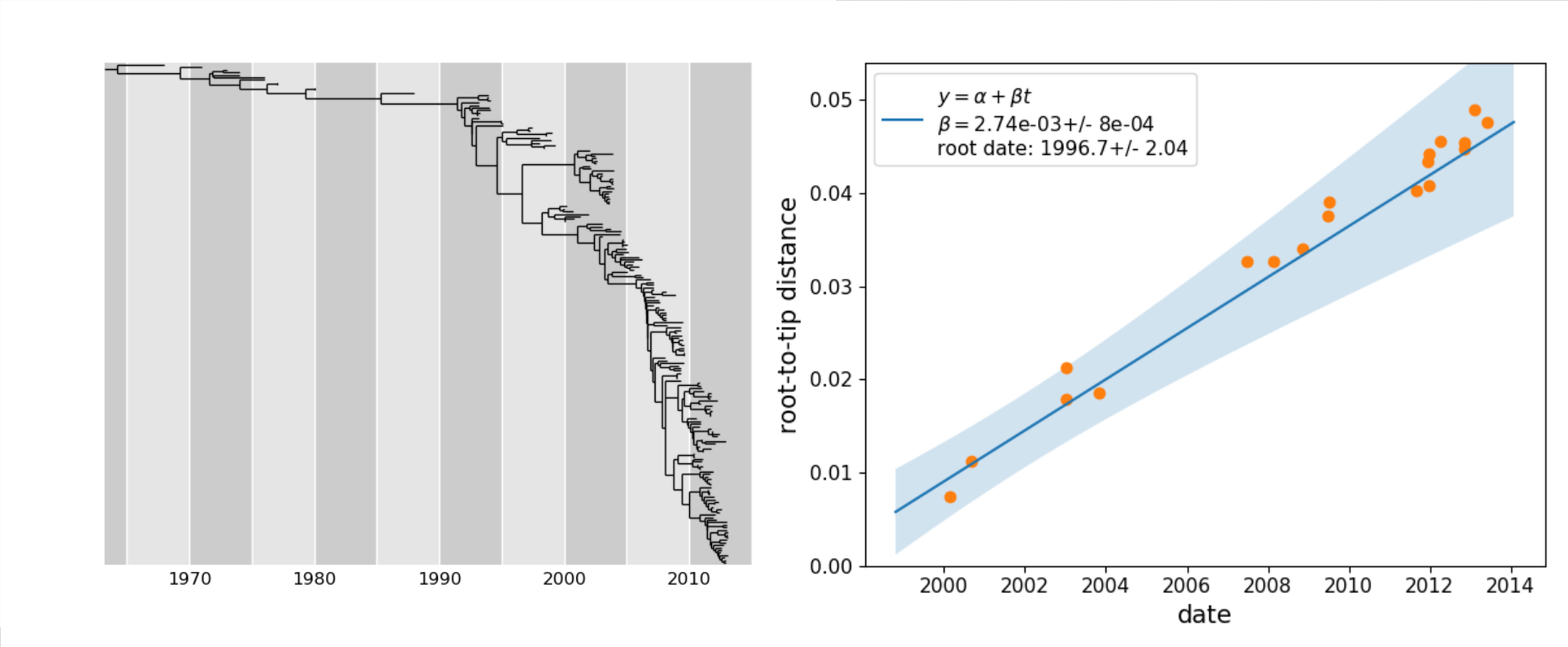
Have a look at our repository with [example data](https://github.com/neherlab/treetime_examples) and the [tutorials](https://treetime.readthedocs.io/en/latest/tutorials.html).
#### Features
* ancestral sequence reconstruction (marginal and joint maximum likelihood)
* molecular clock tree inference (marginal and joint maximum likelihood)
* inference of GTR models
* rerooting to maximize temporal signal and optimize the root-to-tip distance vs time relationship
* simple phylodynamic analysis such as coalescent model fits
* sequence evolution along trees using flexible site specific models.
## Table of contents
* [Installation and prerequisites](#installation-and-prerequisites)
* [Command-line usage](#command-line-usage)
+ [Timetrees](#timetrees)
+ [Rerooting and substitution rate estimation](#rerooting-and-substitution-rate-estimation)
+ [Ancestral sequence reconstruction](#ancestral-sequence-reconstruction)
+ [Homoplasy analysis](#homoplasy-analysis)
+ [Mugration analysis](#mugration-analysis)
+ [Metadata and date format](#metadata-and-date-format)
* [Example scripts](#example-scripts)
* [Related tools](#related-tools)
* [Projects using TreeTime](#projects-using-treetime)
* [Building the documentation](#building-the-documentation)
* [Developer info](#developer-info)
### Installation and prerequisites
TreeTime is compatible with Python 3.7 upwards and is tested on 3.7 to 3.10. It depends on several Python libraries:
* numpy, scipy, pandas: for all kind of mathematical operations as matrix
operations, numerical integration, interpolation, minimization, etc.
* BioPython: for parsing multiple sequence alignments and all phylogenetic
functionality
* matplotlib: optional dependency for plotting
You may install TreeTime and its dependencies by running
```bash
pip install .
```
within this repository.
You can also install TreeTime from PyPi via
```bash
pip install phylo-treetime
```
You might need root privileges for system wide installation. Alternatively, you can simply use it TreeTime locally without installation. In this case, just download and unpack it, and then add the TreeTime folder to your $PYTHONPATH.
### Command-line usage
TreeTime can be used as part of python programs that create and interact with tree time objects. How TreeTime can be used to address typical questions like ancestral sequence reconstruction, rerooting, timetree inference etc is illustrated by a collection of example scripts described below.
In addition, TreeTime can be used from the command line with arguments specifying input data and parameters.
Trees can be read as newick, nexus and phylip files; fasta and phylip are supported alignment formats; metadata and dates can be provided as csv or tsv files, see [below](#metadata-and-date-format) for details.
#### Timetrees
The to infer a timetree, i.e. a phylogenetic tree in which branch length reflect time rather than divergence, TreeTime offers implements the command:
```bash
treetime --aln <input.fasta> --tree <input.nwk> --dates <dates.csv>
```
This command will infer a time tree, ancestral sequences, a GTR model, and optionally confidence intervals and coalescent models.
A detailed explanation is of this command with its various options and examples is available in [the documentation on readthedocs.org](https://treetime.readthedocs.io/en/latest/tutorials/timetree.html).
#### Rerooting and substitution rate estimation
To explore the temporal signal in the data and estimate the substitution rate (instead if full-blown timetree estimation), TreeTime implements a subcommand `clock` that is called as follows
```bash
treetime clock --tree <input.nwk> --aln <input.fasta> --dates <dates.csv> --reroot least-squares
```
The full list if options is available by typing `treetime clock -h`.
Instead of an input alignment, `--sequence-length <L>` can be provided.
Documentation of additional options and examples are available at in [the documentation on readthedocs.org](https://treetime.readthedocs.io/en/latest/tutorials/clock.html).
#### Ancestral sequence reconstruction:
The subcommand
```bash
treetime ancestral --aln input.fasta --tree input.nwk
```
will reconstruct ancestral sequences at internal nodes of the input tree.
The full list if options is available by typing `treetime ancestral -h`.
A detailed explanation of `treetime ancestral` with examples is available at in [the documentation on readthedocs.org](https://treetime.readthedocs.io/en/latest/tutorials/ancestral.html).
#### Homoplasy analysis
Detecting and quantifying homoplasies or recurrent mutations is useful to check for recombination, putative adaptive sites, or contamination.
TreeTime provides a simple command to summarize homoplasies in data
```bash
treetime homoplasy --aln <input.fasta> --tree <input.nwk>
```
The full list if options is available by typing `treetime homoplasy -h`.
Please see [the documentation on readthedocs.org](https://treetime.readthedocs.io/en/latest/tutorials/homoplasy.html) for examples and more documentation.
#### Mugration analysis
Migration between discrete geographic regions, host switching, or other transition between discrete states are often parameterized by time-reversible models analogous to models describing evolution of genome sequences.
Such models are hence often called "mugration" models.
TreeTime GTR model machinery can be used to infer mugration models:
```bash
treetime mugration --tree <input.nwk> --states <states.csv> --attribute <field>
```
where `<field>` is the relevant column in the csv file specifying the metadata `states.csv`, e.g. `<field>=country`.
The full list if options is available by typing `treetime mugration -h`.
Please see [the documentation on readthedocs.org](https://treetime.readthedocs.io/en/latest/tutorials/mugration.html) for examples and more documentation.
#### Metadata and date format
Several of TreeTime commands require the user to specify a file with dates and/or other meta data.
TreeTime assumes these files to by either comma (csv) or tab-separated (tsv) files.
The first line of these files is interpreted as header line specifying the content of the columns.
Each file needs to have at least one column that is named `name`, `accession`, or `strain`.
This column needs to contain the names of each sequence and match the names of taxons in the tree if one is provided.
If more than one of `name`, `accession`, or `strain` is found, TreeTime will use the first.
If the analysis requires dates, at least one column name needs to contain `date` (i.e. `sampling date` is fine).
Again, if multiple hits are found, TreeTime will use the first.
TreeTime will attempt to parse dates in the following way and order
| order | type/format | example | description|
| --- |-------------|---------|------------|
| 1| float | 2017.56 | decimal date |
| 2| [float:float] | [2013.45:2015.56] | decimal date range |
| 3| %Y-%m-%d | 2017-08-25 | calendar date in ISO format |
| 4| %Y-XX-XX | 2017-XX-XX | calendar date missing month and/or day |
### Example scripts
The following scripts illustrate how treetime can be used to solve common problem with short python scripts. They are meant to be used in an interactive ipython environment and run as `run examples/ancestral_inference.py`.
* [`ancestral_inference.py`](https://github.com/neherlab/treetime_examples/tree/master/scripts/ancestral_sequence_inference.py) illustrates how ancestral sequences are inferred and likely mutations are assigned to branches in the tree,
* [`relaxed_clock.py`](https://github.com/neherlab/treetime_examples/tree/master/scripts/relaxed_clock.py) walks the user through the usage of relaxed molecular clock models.
* [`examples/rerooting_and_timetrees.py`](https://github.com/neherlab/treetime_examples/tree/master/scripts/rerooting_and_timetrees.py) illustrates the rerooting and root-to-tip regression scatter plots.
* [`ebola.py`](https://github.com/neherlab/treetime_examples/tree/master/scripts/ebola.py) uses about 300 sequences from the 2014-2015 Ebola virus outbreak to infer a timetree. This example takes a few minutes to run.
HTML documentation of the different classes and function is available at [here](https://treetime.biozentrum.unibas.ch/doc).
### Related tools
There are several other tools which estimate molecular clock phylogenies.
* [Beast](http://beast.bio.ed.ac.uk/) relies on the MCMC-type sampling of trees. It is hence rather slow for large data sets. But BEAST allows the flexible inclusion of prior distributions, complex evolutionary models, and estimation of parameters.
* [Least-Square-Dating](http://www.atgc-montpellier.fr/LSD/) (LSD) emphasizes speed (it scales as O(N) as **TreeTime**), but provides limited scope for customization.
* [treedater](https://github.com/emvolz/treedater) by Eric Volz and Simon Frost is an R package that implements time tree estimation and supports relaxed clocks.
### Projects using TreeTime
* TreeTime is an integral part of the [nextstrain.org](http://nextstrain.org) project to track and analyze viral sequence data in real time.
* [panX](http://pangenome.de) uses TreeTime for ancestral reconstructions and inference of gene gain-loss patterns.
### Building the documentation
The API documentation for the TreeTime package is generated created with Sphinx. The source code for the documentaiton is located in doc folder.
- sphinx-build to generate static html pages from source. Installed as
```bash
pip install Sphinx
```
After required packages are installed, navigate to doc directory, and build the docs by typing:
```bash
make html
```
Instead of html, another target as `latex` or `epub` can be specified to build the docs in the desired format.
#### Requirements
To build the documentation, sphinx-build tool should be installed. The doc pages are using basicstrap html theme to have the same design as the TreeTime web server. Therefore, the basicstrap theme should be also available in the system.
### Developer info
- Copyright and License: Pavel Sagulenko, Emma Hodcroft, and Richard Neher, MIT Licence
- References
* [TreeTime: Maximum-likelihood phylodynamic analysis](https://academic.oup.com/ve/article/4/1/vex042/4794731) by Pavel Sagulenko, Vadim Puller and Richard A Neher. Virus Evolution.
* [NextStrain: real-time tracking of pathogen evolution](https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/bty407/5001388) by James Hadfield et al. Bioinformatics.
Raw data
{
"_id": null,
"home_page": "https://github.com/neherlab/treetime",
"name": "phylo-treetime",
"maintainer": "",
"docs_url": null,
"requires_python": "",
"maintainer_email": "",
"keywords": "Time-stamped phylogenies,phylogeography,virus evolution",
"author": "Pavel Sagulenko, Emma Hodcroft, and Richard Neher",
"author_email": "richard.neher@unibas.ch",
"download_url": "https://files.pythonhosted.org/packages/98/da/0625de3eb3bb4166d389154d122fee3f3cf405a3f57ed0a46be596d8fd2e/phylo-treetime-0.11.2.tar.gz",
"platform": null,
"description": "[](https://github.com/neherlab/treetime/actions/workflows/ci.yml)\n[](https://anaconda.org/bioconda/treetime)\n[](https://treetime.readthedocs.io/en/latest/)\n\n## TreeTime: maximum likelihood dating and ancestral sequence inference\n\n### Overview\n\nTreeTime provides routines for ancestral sequence reconstruction and inference of molecular-clock phylogenies, i.e., a tree where all branches are scaled such that the positions of terminal nodes correspond to their sampling times and internal nodes are placed at the most likely time of divergence.\n\nTo optimize the likelihood of time-scaled phylogenies, TreeTime uses an iterative approach that first infers ancestral sequences given the branch length of the tree (assuming the branch length of the input tree is in units of average number of nucleotide or protein substitutions per site in the sequence). After infering ancestral sequences TreeTime optimizes the positions of unconstrained nodes on the time axis, and then repeats this cycle.\nThe only topology optimization are (optional) resolution of polytomies in a way that is most (approximately) consistent with the sampling time constraints on the tree.\nThe package is designed to be used as a stand-alone tool on the command-line or as a library used in larger phylogenetic analysis work-flows.\n[The documentation of TreeTime is hosted on readthedocs.org](https://treetime.readthedocs.io/en/latest/).\n\nIn addition to scripting TreeTime or using it via the command-line, there is also a small web server at [treetime.ch](https://treetime.biozentrum.unibas.ch/).\n\n\n\nHave a look at our repository with [example data](https://github.com/neherlab/treetime_examples) and the [tutorials](https://treetime.readthedocs.io/en/latest/tutorials.html).\n\n#### Features\n* ancestral sequence reconstruction (marginal and joint maximum likelihood)\n* molecular clock tree inference (marginal and joint maximum likelihood)\n* inference of GTR models\n* rerooting to maximize temporal signal and optimize the root-to-tip distance vs time relationship\n* simple phylodynamic analysis such as coalescent model fits\n* sequence evolution along trees using flexible site specific models.\n\n## Table of contents\n * [Installation and prerequisites](#installation-and-prerequisites)\n * [Command-line usage](#command-line-usage)\n + [Timetrees](#timetrees)\n + [Rerooting and substitution rate estimation](#rerooting-and-substitution-rate-estimation)\n + [Ancestral sequence reconstruction](#ancestral-sequence-reconstruction)\n + [Homoplasy analysis](#homoplasy-analysis)\n + [Mugration analysis](#mugration-analysis)\n + [Metadata and date format](#metadata-and-date-format)\n * [Example scripts](#example-scripts)\n * [Related tools](#related-tools)\n * [Projects using TreeTime](#projects-using-treetime)\n * [Building the documentation](#building-the-documentation)\n * [Developer info](#developer-info)\n\n\n\n### Installation and prerequisites\n\nTreeTime is compatible with Python 3.7 upwards and is tested on 3.7 to 3.10. It depends on several Python libraries:\n\n* numpy, scipy, pandas: for all kind of mathematical operations as matrix\n operations, numerical integration, interpolation, minimization, etc.\n\n* BioPython: for parsing multiple sequence alignments and all phylogenetic\n functionality\n\n* matplotlib: optional dependency for plotting\n\nYou may install TreeTime and its dependencies by running\n\n```bash\n pip install .\n```\nwithin this repository.\nYou can also install TreeTime from PyPi via\n```bash\n pip install phylo-treetime\n```\n\nYou might need root privileges for system wide installation. Alternatively, you can simply use it TreeTime locally without installation. In this case, just download and unpack it, and then add the TreeTime folder to your $PYTHONPATH.\n\n\n### Command-line usage\nTreeTime can be used as part of python programs that create and interact with tree time objects. How TreeTime can be used to address typical questions like ancestral sequence reconstruction, rerooting, timetree inference etc is illustrated by a collection of example scripts described below.\n\nIn addition, TreeTime can be used from the command line with arguments specifying input data and parameters.\nTrees can be read as newick, nexus and phylip files; fasta and phylip are supported alignment formats; metadata and dates can be provided as csv or tsv files, see [below](#metadata-and-date-format) for details.\n\n#### Timetrees\nThe to infer a timetree, i.e. a phylogenetic tree in which branch length reflect time rather than divergence, TreeTime offers implements the command:\n```bash\n treetime --aln <input.fasta> --tree <input.nwk> --dates <dates.csv>\n```\nThis command will infer a time tree, ancestral sequences, a GTR model, and optionally confidence intervals and coalescent models.\nA detailed explanation is of this command with its various options and examples is available in [the documentation on readthedocs.org](https://treetime.readthedocs.io/en/latest/tutorials/timetree.html).\n\n\n#### Rerooting and substitution rate estimation\nTo explore the temporal signal in the data and estimate the substitution rate (instead if full-blown timetree estimation), TreeTime implements a subcommand `clock` that is called as follows\n```bash\n treetime clock --tree <input.nwk> --aln <input.fasta> --dates <dates.csv> --reroot least-squares\n```\nThe full list if options is available by typing `treetime clock -h`.\nInstead of an input alignment, `--sequence-length <L>` can be provided.\nDocumentation of additional options and examples are available at in [the documentation on readthedocs.org](https://treetime.readthedocs.io/en/latest/tutorials/clock.html).\n\n\n#### Ancestral sequence reconstruction:\nThe subcommand\n```bash\n treetime ancestral --aln input.fasta --tree input.nwk\n```\nwill reconstruct ancestral sequences at internal nodes of the input tree.\nThe full list if options is available by typing `treetime ancestral -h`.\nA detailed explanation of `treetime ancestral` with examples is available at in [the documentation on readthedocs.org](https://treetime.readthedocs.io/en/latest/tutorials/ancestral.html).\n\n#### Homoplasy analysis\nDetecting and quantifying homoplasies or recurrent mutations is useful to check for recombination, putative adaptive sites, or contamination.\nTreeTime provides a simple command to summarize homoplasies in data\n```bash\n treetime homoplasy --aln <input.fasta> --tree <input.nwk>\n```\nThe full list if options is available by typing `treetime homoplasy -h`.\nPlease see [the documentation on readthedocs.org](https://treetime.readthedocs.io/en/latest/tutorials/homoplasy.html) for examples and more documentation.\n\n#### Mugration analysis\nMigration between discrete geographic regions, host switching, or other transition between discrete states are often parameterized by time-reversible models analogous to models describing evolution of genome sequences.\nSuch models are hence often called \"mugration\" models.\nTreeTime GTR model machinery can be used to infer mugration models:\n```bash\n treetime mugration --tree <input.nwk> --states <states.csv> --attribute <field>\n```\nwhere `<field>` is the relevant column in the csv file specifying the metadata `states.csv`, e.g. `<field>=country`.\nThe full list if options is available by typing `treetime mugration -h`.\nPlease see [the documentation on readthedocs.org](https://treetime.readthedocs.io/en/latest/tutorials/mugration.html) for examples and more documentation.\n\n#### Metadata and date format\nSeveral of TreeTime commands require the user to specify a file with dates and/or other meta data.\nTreeTime assumes these files to by either comma (csv) or tab-separated (tsv) files.\nThe first line of these files is interpreted as header line specifying the content of the columns.\nEach file needs to have at least one column that is named `name`, `accession`, or `strain`.\nThis column needs to contain the names of each sequence and match the names of taxons in the tree if one is provided.\nIf more than one of `name`, `accession`, or `strain` is found, TreeTime will use the first.\n\nIf the analysis requires dates, at least one column name needs to contain `date` (i.e. `sampling date` is fine).\nAgain, if multiple hits are found, TreeTime will use the first.\nTreeTime will attempt to parse dates in the following way and order\n\n| order | type/format | example | description|\n| --- |-------------|---------|------------|\n| 1| float | 2017.56 | decimal date |\n| 2| [float:float] | [2013.45:2015.56] | decimal date range |\n| 3| %Y-%m-%d | 2017-08-25 | calendar date in ISO format |\n| 4| %Y-XX-XX | 2017-XX-XX | calendar date missing month and/or day |\n\n\n### Example scripts\nThe following scripts illustrate how treetime can be used to solve common problem with short python scripts. They are meant to be used in an interactive ipython environment and run as `run examples/ancestral_inference.py`.\n * [`ancestral_inference.py`](https://github.com/neherlab/treetime_examples/tree/master/scripts/ancestral_sequence_inference.py) illustrates how ancestral sequences are inferred and likely mutations are assigned to branches in the tree,\n * [`relaxed_clock.py`](https://github.com/neherlab/treetime_examples/tree/master/scripts/relaxed_clock.py) walks the user through the usage of relaxed molecular clock models.\n * [`examples/rerooting_and_timetrees.py`](https://github.com/neherlab/treetime_examples/tree/master/scripts/rerooting_and_timetrees.py) illustrates the rerooting and root-to-tip regression scatter plots.\n * [`ebola.py`](https://github.com/neherlab/treetime_examples/tree/master/scripts/ebola.py) uses about 300 sequences from the 2014-2015 Ebola virus outbreak to infer a timetree. This example takes a few minutes to run.\n\nHTML documentation of the different classes and function is available at [here](https://treetime.biozentrum.unibas.ch/doc).\n\n### Related tools\n\nThere are several other tools which estimate molecular clock phylogenies.\n* [Beast](http://beast.bio.ed.ac.uk/) relies on the MCMC-type sampling of trees. It is hence rather slow for large data sets. But BEAST allows the flexible inclusion of prior distributions, complex evolutionary models, and estimation of parameters.\n* [Least-Square-Dating](http://www.atgc-montpellier.fr/LSD/) (LSD) emphasizes speed (it scales as O(N) as **TreeTime**), but provides limited scope for customization.\n* [treedater](https://github.com/emvolz/treedater) by Eric Volz and Simon Frost is an R package that implements time tree estimation and supports relaxed clocks.\n\n### Projects using TreeTime\n\n * TreeTime is an integral part of the [nextstrain.org](http://nextstrain.org) project to track and analyze viral sequence data in real time.\n * [panX](http://pangenome.de) uses TreeTime for ancestral reconstructions and inference of gene gain-loss patterns.\n\n\n### Building the documentation\n\nThe API documentation for the TreeTime package is generated created with Sphinx. The source code for the documentaiton is located in doc folder.\n\n - sphinx-build to generate static html pages from source. Installed as\n\n ```bash\n pip install Sphinx\n ```\n\nAfter required packages are installed, navigate to doc directory, and build the docs by typing:\n\n```bash\nmake html\n```\n\nInstead of html, another target as `latex` or `epub` can be specified to build the docs in the desired format.\n\n\n#### Requirements\n\nTo build the documentation, sphinx-build tool should be installed. The doc pages are using basicstrap html theme to have the same design as the TreeTime web server. Therefore, the basicstrap theme should be also available in the system.\n\n\n### Developer info\n\n - Copyright and License: Pavel Sagulenko, Emma Hodcroft, and Richard Neher, MIT Licence\n - References\n * [TreeTime: Maximum-likelihood phylodynamic analysis](https://academic.oup.com/ve/article/4/1/vex042/4794731) by Pavel Sagulenko, Vadim Puller and Richard A Neher. Virus Evolution.\n * [NextStrain: real-time tracking of pathogen evolution](https://academic.oup.com/bioinformatics/advance-article/doi/10.1093/bioinformatics/bty407/5001388) by James Hadfield et al. Bioinformatics.\n\n",
"bugtrack_url": null,
"license": "MIT",
"summary": "Maximum-likelihood phylodynamic inference",
"version": "0.11.2",
"project_urls": {
"Homepage": "https://github.com/neherlab/treetime"
},
"split_keywords": [
"time-stamped phylogenies",
"phylogeography",
"virus evolution"
],
"urls": [
{
"comment_text": "",
"digests": {
"blake2b_256": "d936e86c7df63fd2441c1c32693ca6e82af093c7e64b56436231877c036d3d23",
"md5": "d341d91a0f956122960a3d979ae6f08a",
"sha256": "1573bbb3c861577f3021c9ac3c69de1ea8e01ac4e4c349a48e0a5f58f9852a5b"
},
"downloads": -1,
"filename": "phylo_treetime-0.11.2-py3-none-any.whl",
"has_sig": false,
"md5_digest": "d341d91a0f956122960a3d979ae6f08a",
"packagetype": "bdist_wheel",
"python_version": "py3",
"requires_python": null,
"size": 148297,
"upload_time": "2024-01-22T09:53:28",
"upload_time_iso_8601": "2024-01-22T09:53:28.189591Z",
"url": "https://files.pythonhosted.org/packages/d9/36/e86c7df63fd2441c1c32693ca6e82af093c7e64b56436231877c036d3d23/phylo_treetime-0.11.2-py3-none-any.whl",
"yanked": false,
"yanked_reason": null
},
{
"comment_text": "",
"digests": {
"blake2b_256": "98da0625de3eb3bb4166d389154d122fee3f3cf405a3f57ed0a46be596d8fd2e",
"md5": "caeb6342f30adb4a2d6774f553a6d317",
"sha256": "2f85dfd13f7addbdce35bf0eaa7f994c6e2065cf992d11bff01644b1ae6341d5"
},
"downloads": -1,
"filename": "phylo-treetime-0.11.2.tar.gz",
"has_sig": false,
"md5_digest": "caeb6342f30adb4a2d6774f553a6d317",
"packagetype": "sdist",
"python_version": "source",
"requires_python": null,
"size": 1298792,
"upload_time": "2024-01-22T09:53:31",
"upload_time_iso_8601": "2024-01-22T09:53:31.125717Z",
"url": "https://files.pythonhosted.org/packages/98/da/0625de3eb3bb4166d389154d122fee3f3cf405a3f57ed0a46be596d8fd2e/phylo-treetime-0.11.2.tar.gz",
"yanked": false,
"yanked_reason": null
}
],
"upload_time": "2024-01-22 09:53:31",
"github": true,
"gitlab": false,
"bitbucket": false,
"codeberg": false,
"github_user": "neherlab",
"github_project": "treetime",
"travis_ci": false,
"coveralls": false,
"github_actions": true,
"lcname": "phylo-treetime"
}

