| Name | Dans-Diffraction JSON |
| Version |
3.3.2
 JSON
JSON |
| download |
| home_page | https://github.com/DanPorter/Dans_Diffraction |
| Summary | Generate diffracted intensities from crystals |
| upload_time | 2024-11-21 19:23:02 |
| maintainer | None |
| docs_url | None |
| author | Dan Porter |
| requires_python | >=3.7 |
| license | Apache License Version 2.0, January 2004 http://www.apache.org/licenses/ TERMS AND CONDITIONS FOR USE, REPRODUCTION, AND DISTRIBUTION 1. Definitions. "License" shall mean the terms and conditions for use, reproduction, and distribution as defined by Sections 1 through 9 of this document. "Licensor" shall mean the copyright owner or entity authorized by the copyright owner that is granting the License. "Legal Entity" shall mean the union of the acting entity and all other entities that control, are controlled by, or are under common control with that entity. For the purposes of this definition, "control" means (i) the power, direct or indirect, to cause the direction or management of such entity, whether by contract or otherwise, or (ii) ownership of fifty percent (50%) or more of the outstanding shares, or (iii) beneficial ownership of such entity. "You" (or "Your") shall mean an individual or Legal Entity exercising permissions granted by this License. "Source" form shall mean the preferred form for making modifications, including but not limited to software source code, documentation source, and configuration files. "Object" form shall mean any form resulting from mechanical transformation or translation of a Source form, including but not limited to compiled object code, generated documentation, and conversions to other media types. "Work" shall mean the work of authorship, whether in Source or Object form, made available under the License, as indicated by a copyright notice that is included in or attached to the work (an example is provided in the Appendix below). "Derivative Works" shall mean any work, whether in Source or Object form, that is based on (or derived from) the Work and for which the editorial revisions, annotations, elaborations, or other modifications represent, as a whole, an original work of authorship. For the purposes of this License, Derivative Works shall not include works that remain separable from, or merely link (or bind by name) to the interfaces of, the Work and Derivative Works thereof. "Contribution" shall mean any work of authorship, including the original version of the Work and any modifications or additions to that Work or Derivative Works thereof, that is intentionally submitted to Licensor for inclusion in the Work by the copyright owner or by an individual or Legal Entity authorized to submit on behalf of the copyright owner. For the purposes of this definition, "submitted" means any form of electronic, verbal, or written communication sent to the Licensor or its representatives, including but not limited to communication on electronic mailing lists, source code control systems, and issue tracking systems that are managed by, or on behalf of, the Licensor for the purpose of discussing and improving the Work, but excluding communication that is conspicuously marked or otherwise designated in writing by the copyright owner as "Not a Contribution." "Contributor" shall mean Licensor and any individual or Legal Entity on behalf of whom a Contribution has been received by Licensor and subsequently incorporated within the Work. 2. Grant of Copyright License. Subject to the terms and conditions of this License, each Contributor hereby grants to You a perpetual, worldwide, non-exclusive, no-charge, royalty-free, irrevocable copyright license to reproduce, prepare Derivative Works of, publicly display, publicly perform, sublicense, and distribute the Work and such Derivative Works in Source or Object form. 3. Grant of Patent License. Subject to the terms and conditions of this License, each Contributor hereby grants to You a perpetual, worldwide, non-exclusive, no-charge, royalty-free, irrevocable (except as stated in this section) patent license to make, have made, use, offer to sell, sell, import, and otherwise transfer the Work, where such license applies only to those patent claims licensable by such Contributor that are necessarily infringed by their Contribution(s) alone or by combination of their Contribution(s) with the Work to which such Contribution(s) was submitted. If You institute patent litigation against any entity (including a cross-claim or counterclaim in a lawsuit) alleging that the Work or a Contribution incorporated within the Work constitutes direct or contributory patent infringement, then any patent licenses granted to You under this License for that Work shall terminate as of the date such litigation is filed. 4. Redistribution. You may reproduce and distribute copies of the Work or Derivative Works thereof in any medium, with or without modifications, and in Source or Object form, provided that You meet the following conditions: (a) You must give any other recipients of the Work or Derivative Works a copy of this License; and (b) You must cause any modified files to carry prominent notices stating that You changed the files; and (c) You must retain, in the Source form of any Derivative Works that You distribute, all copyright, patent, trademark, and attribution notices from the Source form of the Work, excluding those notices that do not pertain to any part of the Derivative Works; and (d) If the Work includes a "NOTICE" text file as part of its distribution, then any Derivative Works that You distribute must include a readable copy of the attribution notices contained within such NOTICE file, excluding those notices that do not pertain to any part of the Derivative Works, in at least one of the following places: within a NOTICE text file distributed as part of the Derivative Works; within the Source form or documentation, if provided along with the Derivative Works; or, within a display generated by the Derivative Works, if and wherever such third-party notices normally appear. The contents of the NOTICE file are for informational purposes only and do not modify the License. You may add Your own attribution notices within Derivative Works that You distribute, alongside or as an addendum to the NOTICE text from the Work, provided that such additional attribution notices cannot be construed as modifying the License. You may add Your own copyright statement to Your modifications and may provide additional or different license terms and conditions for use, reproduction, or distribution of Your modifications, or for any such Derivative Works as a whole, provided Your use, reproduction, and distribution of the Work otherwise complies with the conditions stated in this License. 5. Submission of Contributions. Unless You explicitly state otherwise, any Contribution intentionally submitted for inclusion in the Work by You to the Licensor shall be under the terms and conditions of this License, without any additional terms or conditions. Notwithstanding the above, nothing herein shall supersede or modify the terms of any separate license agreement you may have executed with Licensor regarding such Contributions. 6. Trademarks. This License does not grant permission to use the trade names, trademarks, service marks, or product names of the Licensor, except as required for reasonable and customary use in describing the origin of the Work and reproducing the content of the NOTICE file. 7. Disclaimer of Warranty. Unless required by applicable law or agreed to in writing, Licensor provides the Work (and each Contributor provides its Contributions) on an "AS IS" BASIS, WITHOUT WARRANTIES OR CONDITIONS OF ANY KIND, either express or implied, including, without limitation, any warranties or conditions of TITLE, NON-INFRINGEMENT, MERCHANTABILITY, or FITNESS FOR A PARTICULAR PURPOSE. You are solely responsible for determining the appropriateness of using or redistributing the Work and assume any risks associated with Your exercise of permissions under this License. 8. Limitation of Liability. In no event and under no legal theory, whether in tort (including negligence), contract, or otherwise, unless required by applicable law (such as deliberate and grossly negligent acts) or agreed to in writing, shall any Contributor be liable to You for damages, including any direct, indirect, special, incidental, or consequential damages of any character arising as a result of this License or out of the use or inability to use the Work (including but not limited to damages for loss of goodwill, work stoppage, computer failure or malfunction, or any and all other commercial damages or losses), even if such Contributor has been advised of the possibility of such damages. 9. Accepting Warranty or Additional Liability. While redistributing the Work or Derivative Works thereof, You may choose to offer, and charge a fee for, acceptance of support, warranty, indemnity, or other liability obligations and/or rights consistent with this License. However, in accepting such obligations, You may act only on Your own behalf and on Your sole responsibility, not on behalf of any other Contributor, and only if You agree to indemnify, defend, and hold each Contributor harmless for any liability incurred by, or claims asserted against, such Contributor by reason of your accepting any such warranty or additional liability. END OF TERMS AND CONDITIONS APPENDIX: How to apply the Apache License to your work. To apply the Apache License to your work, attach the following boilerplate notice, with the fields enclosed by brackets "[]" replaced with your own identifying information. (Don't include the brackets!) The text should be enclosed in the appropriate comment syntax for the file format. We also recommend that a file or class name and description of purpose be included on the same "printed page" as the copyright notice for easier identification within third-party archives. Copyright [yyyy] [name of copyright owner] Licensed under the Apache License, Version 2.0 (the "License"); you may not use this file except in compliance with the License. You may obtain a copy of the License at http://www.apache.org/licenses/LICENSE-2.0 Unless required by applicable law or agreed to in writing, software distributed under the License is distributed on an "AS IS" BASIS, WITHOUT WARRANTIES OR CONDITIONS OF ANY KIND, either express or implied. See the License for the specific language governing permissions and limitations under the License. |
| keywords |
crystal
cif
diffraction
crystallography
science
x-ray
neutron
resonant
magnetic
magnetism
multiple scattering
fdmnes
super structure
spacegroup
space group
diffractometer
|
| VCS |
 |
| bugtrack_url |
|
| requirements |
No requirements were recorded.
|
| Travis-CI |
No Travis.
|
| coveralls test coverage |
No coveralls.
|
# Dans_Diffraction
Reads crystallographic cif files, calculates crystal properties and simulates diffraction.
**Version 3.3**
[](https://doi.org/10.5281/zenodo.8106031)
[](https://mybinder.org/v2/gh/DanPorter/Dans_Diffraction/master?labpath=Dans_Diffraction.ipynb)
[](https://github.com/DanPorter/Dans_Diffraction)
By Dan Porter, Diamond Light Source
2024
#### TL;DR:
```text
$ ipython -i -m Dans_Diffraction
OR
$ ipython -m Dans_Diffraction gui
```
```python
"""Python Sctipt"""
import Dans_Diffraction as dif
xtl = dif.Crystal('some_file.cif')
print(xtl) # print Crystal structure parameters
# Print reflection list:
print(xtl.Scatter.print_all_reflections(energy_kev=5))
# Plot Powder pattern:
xtl.Plot.simulate_powder(energy_kev=8)
plt.show()
# Start graphical user interface:
xtl.start_gui()
```
Full code documentation available [here](https://danporter.github.io/Dans_Diffraction/).
Try it out on [mybinder!](https://mybinder.org/v2/gh/DanPorter/Dans_Diffraction/master?labpath=Dans_Diffraction.ipynb)
For comments, queries or bugs - email [dan.porter@diamond.ac.uk](mailto:dan.porter@diamond.ac.uk)
**Citation:** If you use this code (great!), please cite the published DOI: [10.5281/zenodo.8106031](https://doi.org/10.5281/zenodo.8106031)
# Installation
**Requirements:**
Python 3+ with packages: *Numpy*, *Matplotlib*, *Tkinter*.
BuiltIn packages used: *sys*, *os*, *re*, *glob*, *warnings*, *json*, *itertools*
Install stable version from PyPI:
```text
$ python -m pip install Dans-Diffraction
```
Or, install the latest version direct from GitHub:
```text
$ python -m pip install git+https://github.com/DanPorter/Dans_Diffraction.git
```
Or, Download the latest version from GitHub (with examples!):
```text
$ git clone https://github.com/DanPorter/Dans_Diffraction.git
$ cd Dans_Diffraction
$ python -m pip install .
```
# Operation
From version 3.2, installing Dans_Diffraction will include a run script for the gui:
```text
$ dansdiffraction
```
Alternatively, Dans_Diffraction is best run within an interactive python environment:
```text
$ ipython -i -m Dans_Diffraction
```
Dans_Diffraction can also be run in scripts as an import, example scripts are provided in the [Examples](https://github.com/DanPorter/Dans_Diffraction/blob/master/Examples) folder.
### Read CIF file
```python
import Dans_Diffraction as dif
xtl = dif.Crystal('some_file.cif')
xtl.info() # print Crystal structure parameters
help(xtl) # all functions (nearly!) are documented
```
### Alter atomic positions
```python
xtl.Cell.latt([2.85,2.85,10.8,90,90,120]) # set lattice parameters
xtl.Atoms.info() # Print Symmetric positions
xtl.Structure.info() # Print All positions in P1 symmetry (same structure and functions as xtl.Atoms)
# Symmetric positions
xtl.Atoms.changeatom(idx=0, u=0, v=0, w=0, type='Co', label='Co1')
xtl.Atoms.addatom(idx=0, u=0, v=0, w=0, type='Co', label='Co1')
# After adding or changing an atom in the Atoms class, re-generate the full structure using symmetry arguments:
xtl.generate_lattice()
# Full atomic structure in P1 symmetry
xtl.Structure.changeatom(idx=0, u=0, v=0, w=0, type='Co', label='Co1')
xtl.Structure.addatom(idx=0, u=0, v=0, w=0, type='Co', label='Co1')
# Plot crystal Structure
xtl.Plot.plot_crystal() # 3D plot
xtl.Plot.plot_layers() # 2D plot for layered materials
```

### Alter crystal symmetry
```python
xtl.Symmetry.info() # print symmetry arguments
xtl.Symmetry.addsym('x,y,z+1/2') # adds single symmetry operation
xtl.Symmetry.changesym(0, 'x,y,z+1/4')
xtl.Symmetry.load_spacegroup(194) # replaces current symmetry operations
# After adding or changing symmetry operations, regengerate the symmetry matrices
xtl.Symmetry.generate_matrices()
```
### Save structure as CIF
Lattice parameters, crystal structure and symmetry operations will be saved to the CIF.
If magnetic moments are defined, magnetic symmetry operations and moments will also be saved
and format changed to "*.mcif".
```python
xtl.write_cif('edited file.cif')
```
### Calculate Structure Factors
X-ray or neutron structure factors/ intensities are calculated based on the full unit cell structure, including atomic
form-factors (x-rays) or coherent scattering lengths (neutrons).
```python
# Choose scattering options (see help(xtl.Scatter.setup_scatter))
xtl.Scatter.setup_scatter(scattering_type='x-ray', energy_keV=8.0)
# Allowed radiation types:
# 'xray','neutron','xray magnetic','neutron magnetic','xray resonant'
xtl.Scatter.print_all_reflections() # Returns formated string of all allowed reflections
inten = xtl.Scatter.intensity([h,k,l]) # Returns intensity
twotheta, iten, reflections = xtl.Scatter.powder(units='twotheta')
# Plot Experimental Intensities
xtl.Plot.simulate_powder() # Powder pattern
xtl.Plot.simulate_hk0() # Reciprocal space plane
```

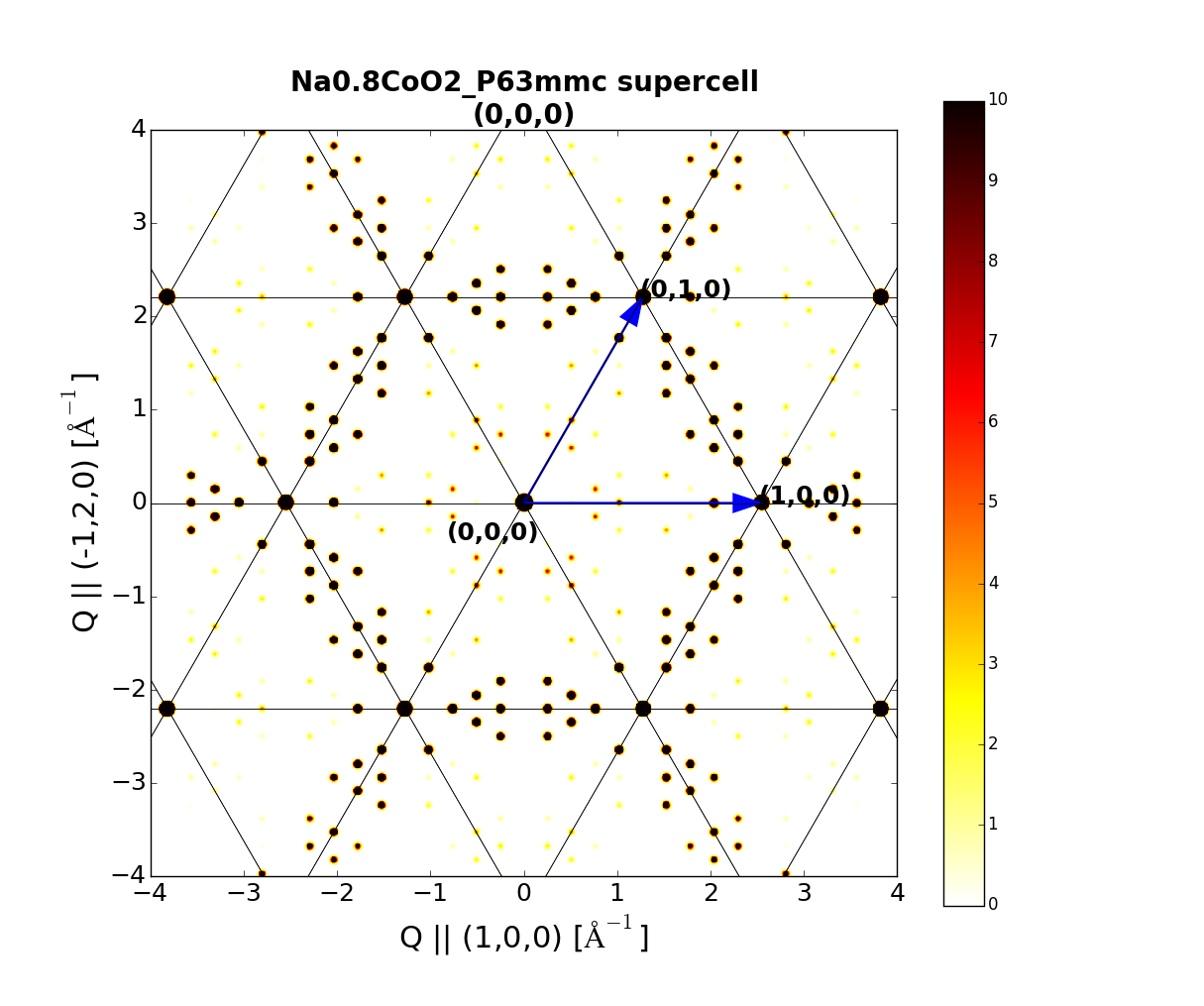
### Magnetic Structrues
*Magnetic structures and scattering are currently in development and shouldn't be treated as accurate!*
Simple magnetic structures can be loaded from magnetic cif (*.mcif) files. Magnetic moments are stored for each atomic
position as a vector. The crystal object has a seperate set of magnetic symmetry operations. Symmetry operations from the
tables of magnetic spacegroups can also be loaded. Only simple magnetic structures are allowed. There must be the same
number of magnetic symmetry operations as crystal symmetry operations and atomic positions can only have single moments
assigned.
```python
xtl = dif.Crystal('some_file.mcif')
xtl.Atoms.mxmymz() # return magnetic moment vectors on each ion
xtl.Symmetry.symmetry_operations_magnetic # magnetic symmetry operations (list of strings)
xtl.Symmetry.print_magnetic_spacegroups() # return str of available magnetic spacegroups, given crystal's spacegroup
xtl.Symmetry.load_magnetic_spacegroup(mag_spg_number) # loads mag. operations given mag. spacegroup number
```
Magnetic scattering is also available for neutrons and x-rays (both resonant and non-resonant), using the appropriate magnetic form-factors.
```python
Imag = xtl.Scatter.magnetic_neutron(HKL=[0,0,3])
Ires = xtl.Scatter.xray_resonant_magnetic(HKL=[0,0,3], energy_kev=2.838, azim_zero=[1, 0, 0], psi=0, polarisation='s-p', F0=0, F1=1, F2=0)
```
### Superstructures
Superstructures can be built using the Superstructure class, requring only a matrix to define the new phase:
```python
su = xtl.generate_superstructure([[2,0,0],[0,2,0],[0,0,1]])
```
Superstucture classes behave like Crystal classes, but have an additional 'Parent' property that references the original
crystal structure and additional behaviours partiular to superstructures. Superstructures loose their parent crystal and
magnetic symmetry, always being defined in P1 symmetry. So su.Atoms == su.Structure.
```python
print(su.parent.info()) # Parent structure
su.P # superstructure matrix
su.superhkl2parent([h, k, l]) # index superstructure hkl with parent cell
su.parenthkl2super([h, k, l]) # index parent hkl with supercell
```
### Multi-phase
Scattering from different crystal structures can be compared using the MultiCrystal class:
```python
xtls = xtl1 + xtl2
xtls.simulate_powder()
```
### Properties
The Crystal class contains a lot of atomic properties that can be exposed in the Properties class:
```python
xtl.Properties.info()
```
Calculated properties include:
- Molecular weight
- Density
- Diamagnetic suscpetibility
- x-ray absorption coefficient, attenuation length, transmission and refractive index
- Molecular charge balance
- Molecular mass fraction
- Atomic orbitals
- Magnetic exchange paths (in progress...)
Properties are calulated using the atomic structure along with atomic data stored in the folder [Dans_Diffraction/data](data).
### Multiple Scattering
Simulations of multiple scattering at different azimuths for a particular energy can be simulated. Based on [code by Dr Gareth Nisbet](https://journals.iucr.org/a/issues/2015/01/00/td5022/).
[](https://doi.org/10.5281/zenodo.12866).
```python
azimuth, intensity = xtl.Scatter.ms_azimuth([h,k,l], energy_kev=8)
```

### Graphical Front End

Start a new GUI, then select a cif file:
```text
$ ipython -i -m Dans_Diffraction gui
```
Or start the GUI from within the interactive console:
```python
dif.start_gui()
```
Using an already generated crystal:
```python
xtl.start_gui()
```
### Diffractometer Simulator

New in version 3.0.0. Simulate a generic detector situated around the crystal sample, with the ability to
control detector location shape and size and lattice orientation.
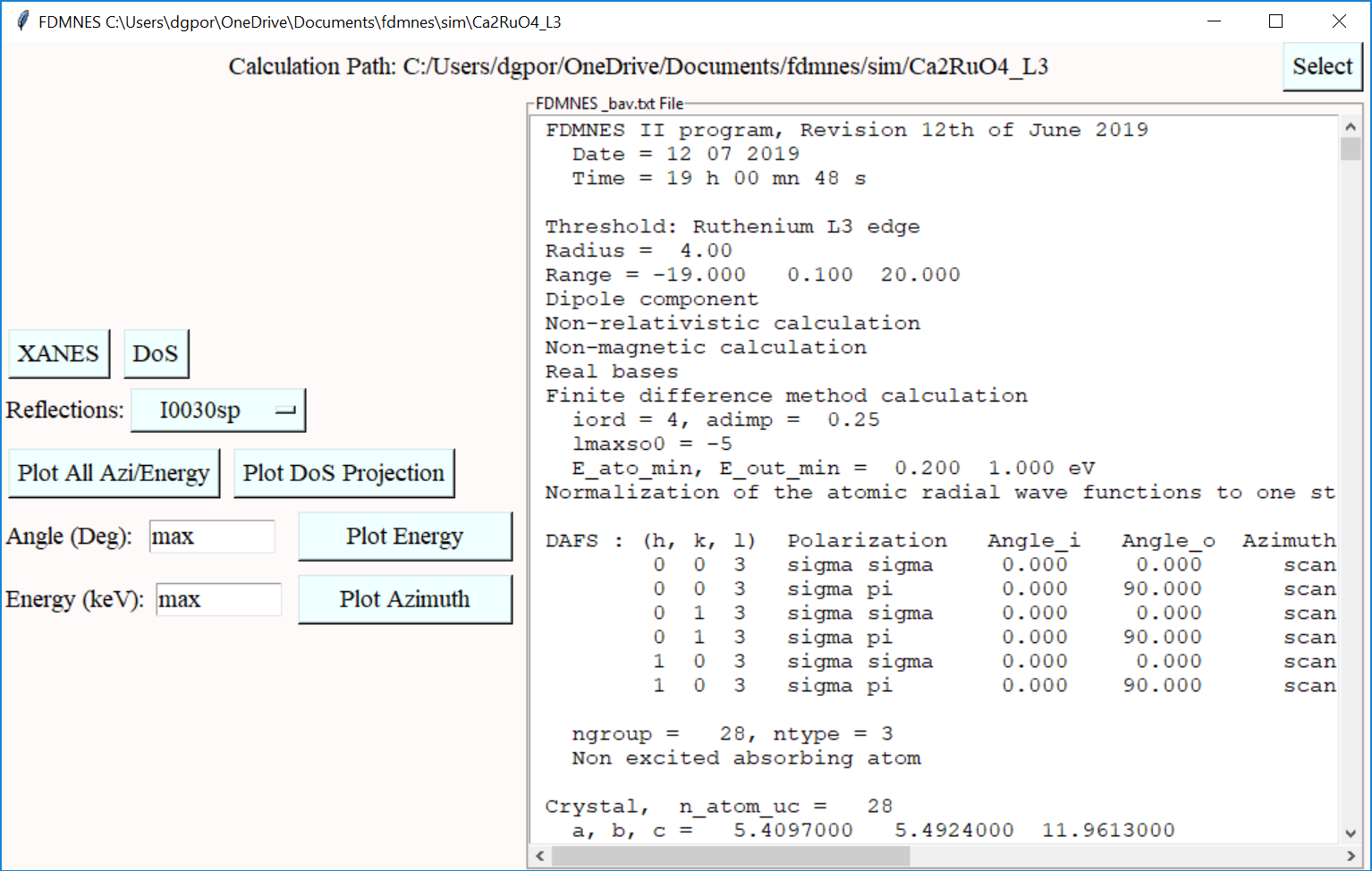
### FDMNES functionality
FDMNES is a powerful tool for simulating resonant x-ray diffraction, created by [Y. Joly and O. Bunau.](https://fdmnes.neel.cnrs.fr/)
The Dans_Diffraction FDMNES class allows for the automatic creation of input files and simple analysis of results.
The following command should be used to activate these features (only needs to be issued once).
```python
dif.activate_fdmnes()
```
Once activated, the FDMNES classes become available.
```python
fdm = dif.Fdmnes(xtl) # Create input files and run FDMNES
fdma = dif.FdmnesAnalysis(output_path, output_name) # Load output files and plot results
```
See class documentation for more information.
Once activated, FDMNES GUI elements become available from the main window, emulating functionality of the classes.

# Acknoledgements
| Date | Thanks to... |
|-------------|-----------------------------------------------------------------------------------------|
| 2018 | Thanks to Hepesu for help with Python3 support and ideas about breaking up calculations |
| Dec 2019 | Thanks to Gareth Nisbet for allowing me to inlude his multiple scattering siumulation |
| April 2020 | Thanks to ChunHai Wang for helpful suggestions in readcif! |
| May 2020 | Thanks to AndreEbel for helpful suggestions on citations |
| Dec 2020 | Thanks to Chris Drozdowski for suggestions about reflection families |
| Jan 2021 | Thanks to aslarsen for suggestions about outputting the structure factor |
| April 2021 | Thanks to Trygve Ræder for suggestions about x-ray scattering factors |
| Feb 2022 | Thanks to Mirko for pointing out the error in two-theta values in Scatter.powder |
| March 2022 | Thanks to yevgenyr for suggesting new peak profiles in Scatter.powder |
| Jan 2023 | Thanks to Anuradha Vibhakar for pointing out the error in f0 + if'-if'' |
| Jan 2023 | Thanks to Andreas Rosnes for testing the installation in jupyterlab |
| May 2023 | Thanks to Carmelo Prestipino for adding electron scattering factors |
| June 2023 | Thanks to Sergio I. Rincon for pointing out the rounding error in Scatter.powder |
| July 2023 | Thanks to asteppke for suggested update to Arrow3D for matplotlib V>3.4 |
| July 2023 | Thanks to Yves Joly for helpful suggestions on FDMNES wrapper |
| Jan 2024 | Thanks to Carmelo Prestipino for adding search_distance and plot_distance |
| April 2024 | Thanks to Innbig for pointing out an issue with liquid crystal simulations |
| May 2024 | Thanks to paul-cares pointing out a silly spelling error in the title! |
Copyright
-----------------------------------------------------------------------------
Copyright 2024 Diamond Light Source Ltd.
Licensed under the Apache License, Version 2.0 (the "License");
you may not use this file except in compliance with the License.
You may obtain a copy of the License at
http://www.apache.org/licenses/LICENSE-2.0
Unless required by applicable law or agreed to in writing, software
distributed under the License is distributed on an "AS IS" BASIS,
WITHOUT WARRANTIES OR CONDITIONS OF ANY KIND, either express or implied.
See the License for the specific language governing permissions and
limitations under the License.
Files in this package covered by this licence:
* classes_crystal.py
* classes_scattering.py
* classes_plotting.py
* classes_properties.py
* classes_multicrystal.py
* classes_orientation.py
* classes_orbitals.py
* functions_general.py
* functions_plotting.py
* functions_scattering.py
* functions_crystallography.py
* tkgui/*.py
Other files are either covered by their own licence or not licenced for other use.
| Dr Daniel G Porter | [dan.porter@diamond.ac.uk](mailto:dan.porter@diamond.ac.uk) |
| ---- | ---- |
| [www.diamond.ac.uk](www.diamond.ac.uk) | Diamond Light Source, Chilton, Didcot, Oxon, OX11 0DE, U.K. |
Raw data
{
"_id": null,
"home_page": "https://github.com/DanPorter/Dans_Diffraction",
"name": "Dans-Diffraction",
"maintainer": null,
"docs_url": null,
"requires_python": ">=3.7",
"maintainer_email": "Dan Porter <d.g.porter@outlook.com>",
"keywords": "crystal, cif, diffraction, crystallography, science, x-ray, neutron, resonant, magnetic, magnetism, multiple scattering, fdmnes, super structure, spacegroup, space group, diffractometer",
"author": "Dan Porter",
"author_email": "Dan Porter <d.g.porter@outlook.com>",
"download_url": "https://files.pythonhosted.org/packages/3a/c5/6bd0f043b1de74f939ff84a0d89f78d4a40b700769ba120c112db73015a7/dans_diffraction-3.3.2.tar.gz",
"platform": null,
"description": "# Dans_Diffraction\r\nReads crystallographic cif files, calculates crystal properties and simulates diffraction.\r\n\r\n**Version 3.3**\r\n\r\n[](https://doi.org/10.5281/zenodo.8106031)\r\n[](https://mybinder.org/v2/gh/DanPorter/Dans_Diffraction/master?labpath=Dans_Diffraction.ipynb) \r\n[](https://github.com/DanPorter/Dans_Diffraction)\r\n\r\n\r\nBy Dan Porter, Diamond Light Source\r\n2024\r\n\r\n#### TL;DR:\r\n```text\r\n$ ipython -i -m Dans_Diffraction\r\nOR\r\n$ ipython -m Dans_Diffraction gui\r\n```\r\n\r\n```python\r\n\"\"\"Python Sctipt\"\"\"\r\nimport Dans_Diffraction as dif\r\nxtl = dif.Crystal('some_file.cif')\r\nprint(xtl) # print Crystal structure parameters\r\n\r\n# Print reflection list:\r\nprint(xtl.Scatter.print_all_reflections(energy_kev=5)) \r\n\r\n# Plot Powder pattern:\r\nxtl.Plot.simulate_powder(energy_kev=8)\r\nplt.show()\r\n\r\n# Start graphical user interface:\r\nxtl.start_gui()\r\n```\r\n\r\nFull code documentation available [here](https://danporter.github.io/Dans_Diffraction/).\r\n\r\nTry it out on [mybinder!](https://mybinder.org/v2/gh/DanPorter/Dans_Diffraction/master?labpath=Dans_Diffraction.ipynb)\r\n\r\nFor comments, queries or bugs - email [dan.porter@diamond.ac.uk](mailto:dan.porter@diamond.ac.uk)\r\n\r\n**Citation:** If you use this code (great!), please cite the published DOI: [10.5281/zenodo.8106031](https://doi.org/10.5281/zenodo.8106031)\r\n\r\n# Installation\r\n**Requirements:** \r\nPython 3+ with packages: *Numpy*, *Matplotlib*, *Tkinter*.\r\nBuiltIn packages used: *sys*, *os*, *re*, *glob*, *warnings*, *json*, *itertools*\r\n\r\nInstall stable version from PyPI:\r\n```text\r\n$ python -m pip install Dans-Diffraction\r\n```\r\n\r\nOr, install the latest version direct from GitHub:\r\n```text\r\n$ python -m pip install git+https://github.com/DanPorter/Dans_Diffraction.git\r\n```\r\n\r\nOr, Download the latest version from GitHub (with examples!):\r\n```text\r\n$ git clone https://github.com/DanPorter/Dans_Diffraction.git\r\n$ cd Dans_Diffraction\r\n$ python -m pip install .\r\n```\r\n\r\n\r\n\r\n# Operation\r\nFrom version 3.2, installing Dans_Diffraction will include a run script for the gui:\r\n```text\r\n$ dansdiffraction\r\n```\r\nAlternatively, Dans_Diffraction is best run within an interactive python environment:\r\n```text\r\n$ ipython -i -m Dans_Diffraction\r\n```\r\n\r\nDans_Diffraction can also be run in scripts as an import, example scripts are provided in the [Examples](https://github.com/DanPorter/Dans_Diffraction/blob/master/Examples) folder.\r\n### Read CIF file\r\n```python\r\nimport Dans_Diffraction as dif\r\nxtl = dif.Crystal('some_file.cif')\r\nxtl.info() # print Crystal structure parameters\r\nhelp(xtl) # all functions (nearly!) are documented\r\n```\r\n\r\n### Alter atomic positions\r\n```python\r\nxtl.Cell.latt([2.85,2.85,10.8,90,90,120]) # set lattice parameters\r\nxtl.Atoms.info() # Print Symmetric positions\r\nxtl.Structure.info() # Print All positions in P1 symmetry (same structure and functions as xtl.Atoms)\r\n# Symmetric positions\r\nxtl.Atoms.changeatom(idx=0, u=0, v=0, w=0, type='Co', label='Co1')\r\nxtl.Atoms.addatom(idx=0, u=0, v=0, w=0, type='Co', label='Co1')\r\n# After adding or changing an atom in the Atoms class, re-generate the full structure using symmetry arguments:\r\nxtl.generate_lattice()\r\n# Full atomic structure in P1 symmetry\r\nxtl.Structure.changeatom(idx=0, u=0, v=0, w=0, type='Co', label='Co1')\r\nxtl.Structure.addatom(idx=0, u=0, v=0, w=0, type='Co', label='Co1')\r\n# Plot crystal Structure\r\nxtl.Plot.plot_crystal() # 3D plot\r\nxtl.Plot.plot_layers() # 2D plot for layered materials\r\n```\r\n\r\n\r\n\r\n### Alter crystal symmetry\r\n```python\r\nxtl.Symmetry.info() # print symmetry arguments\r\nxtl.Symmetry.addsym('x,y,z+1/2') # adds single symmetry operation\r\nxtl.Symmetry.changesym(0, 'x,y,z+1/4')\r\nxtl.Symmetry.load_spacegroup(194) # replaces current symmetry operations\r\n# After adding or changing symmetry operations, regengerate the symmetry matrices\r\nxtl.Symmetry.generate_matrices()\r\n```\r\n\r\n### Save structure as CIF\r\nLattice parameters, crystal structure and symmetry operations will be saved to the CIF.\r\nIf magnetic moments are defined, magnetic symmetry operations and moments will also be saved\r\nand format changed to \"*.mcif\".\r\n```python\r\nxtl.write_cif('edited file.cif')\r\n```\r\n\r\n### Calculate Structure Factors\r\nX-ray or neutron structure factors/ intensities are calculated based on the full unit cell structure, including atomic \r\nform-factors (x-rays) or coherent scattering lengths (neutrons).\r\n```python\r\n# Choose scattering options (see help(xtl.Scatter.setup_scatter))\r\nxtl.Scatter.setup_scatter(scattering_type='x-ray', energy_keV=8.0)\r\n# Allowed radiation types:\r\n# 'xray','neutron','xray magnetic','neutron magnetic','xray resonant'\r\nxtl.Scatter.print_all_reflections() # Returns formated string of all allowed reflections\r\ninten = xtl.Scatter.intensity([h,k,l]) # Returns intensity\r\ntwotheta, iten, reflections = xtl.Scatter.powder(units='twotheta')\r\n# Plot Experimental Intensities\r\nxtl.Plot.simulate_powder() # Powder pattern\r\nxtl.Plot.simulate_hk0() # Reciprocal space plane\r\n```\r\n\r\n\r\n\r\n\r\n### Magnetic Structrues\r\n*Magnetic structures and scattering are currently in development and shouldn't be treated as accurate!*\r\n\r\nSimple magnetic structures can be loaded from magnetic cif (*.mcif) files. Magnetic moments are stored for each atomic \r\nposition as a vector. The crystal object has a seperate set of magnetic symmetry operations. Symmetry operations from the \r\ntables of magnetic spacegroups can also be loaded. Only simple magnetic structures are allowed. There must be the same\r\nnumber of magnetic symmetry operations as crystal symmetry operations and atomic positions can only have single moments\r\nassigned.\r\n```python\r\nxtl = dif.Crystal('some_file.mcif')\r\nxtl.Atoms.mxmymz() # return magnetic moment vectors on each ion\r\nxtl.Symmetry.symmetry_operations_magnetic # magnetic symmetry operations (list of strings)\r\nxtl.Symmetry.print_magnetic_spacegroups() # return str of available magnetic spacegroups, given crystal's spacegroup\r\nxtl.Symmetry.load_magnetic_spacegroup(mag_spg_number) # loads mag. operations given mag. spacegroup number\r\n```\r\nMagnetic scattering is also available for neutrons and x-rays (both resonant and non-resonant), using the appropriate magnetic form-factors.\r\n```python\r\nImag = xtl.Scatter.magnetic_neutron(HKL=[0,0,3])\r\nIres = xtl.Scatter.xray_resonant_magnetic(HKL=[0,0,3], energy_kev=2.838, azim_zero=[1, 0, 0], psi=0, polarisation='s-p', F0=0, F1=1, F2=0)\r\n```\r\n\r\n### Superstructures\r\nSuperstructures can be built using the Superstructure class, requring only a matrix to define the new phase:\r\n```python\r\nsu = xtl.generate_superstructure([[2,0,0],[0,2,0],[0,0,1]])\r\n```\r\n\r\nSuperstucture classes behave like Crystal classes, but have an additional 'Parent' property that references the original \r\ncrystal structure and additional behaviours partiular to superstructures. Superstructures loose their parent crystal and\r\nmagnetic symmetry, always being defined in P1 symmetry. So su.Atoms == su.Structure.\r\n\r\n```python\r\nprint(su.parent.info()) # Parent structure\r\nsu.P # superstructure matrix \r\nsu.superhkl2parent([h, k, l]) # index superstructure hkl with parent cell\r\nsu.parenthkl2super([h, k, l]) # index parent hkl with supercell\r\n```\r\n\r\n### Multi-phase\r\nScattering from different crystal structures can be compared using the MultiCrystal class:\r\n```python\r\nxtls = xtl1 + xtl2\r\nxtls.simulate_powder()\r\n```\r\n\r\n\r\n### Properties\r\nThe Crystal class contains a lot of atomic properties that can be exposed in the Properties class:\r\n```python\r\nxtl.Properties.info()\r\n```\r\n\r\nCalculated properties include:\r\n - Molecular weight\r\n - Density\r\n - Diamagnetic suscpetibility \r\n - x-ray absorption coefficient, attenuation length, transmission and refractive index\r\n - Molecular charge balance\r\n - Molecular mass fraction\r\n - Atomic orbitals\r\n - Magnetic exchange paths (in progress...)\r\n\r\nProperties are calulated using the atomic structure along with atomic data stored in the folder [Dans_Diffraction/data](data).\r\n\r\n\r\n### Multiple Scattering\r\nSimulations of multiple scattering at different azimuths for a particular energy can be simulated. Based on [code by Dr Gareth Nisbet](https://journals.iucr.org/a/issues/2015/01/00/td5022/).\r\n [](https://doi.org/10.5281/zenodo.12866).\r\n\r\n```python\r\nazimuth, intensity = xtl.Scatter.ms_azimuth([h,k,l], energy_kev=8)\r\n```\r\n\r\n\r\n\r\n\r\n### Graphical Front End\r\n\r\n\r\nStart a new GUI, then select a cif file:\r\n```text\r\n$ ipython -i -m Dans_Diffraction gui\r\n```\r\nOr start the GUI from within the interactive console:\r\n```python\r\ndif.start_gui()\r\n```\r\nUsing an already generated crystal:\r\n```python\r\nxtl.start_gui()\r\n```\r\n\r\n### Diffractometer Simulator\r\n\r\n\r\nNew in version 3.0.0. Simulate a generic detector situated around the crystal sample, with the ability to \r\ncontrol detector location shape and size and lattice orientation.\r\n\r\n\r\n### FDMNES functionality\r\nFDMNES is a powerful tool for simulating resonant x-ray diffraction, created by [Y. Joly and O. Bunau.](https://fdmnes.neel.cnrs.fr/)\r\n\r\nThe Dans_Diffraction FDMNES class allows for the automatic creation of input files and simple analysis of results.\r\nThe following command should be used to activate these features (only needs to be issued once). \r\n```python\r\ndif.activate_fdmnes()\r\n```\r\nOnce activated, the FDMNES classes become available.\r\n```python\r\nfdm = dif.Fdmnes(xtl) # Create input files and run FDMNES\r\nfdma = dif.FdmnesAnalysis(output_path, output_name) # Load output files and plot results\r\n```\r\nSee class documentation for more information.\r\n\r\n\r\nOnce activated, FDMNES GUI elements become available from the main window, emulating functionality of the classes.\r\n\r\n\r\n\r\n\r\n\r\n# Acknoledgements\r\n| Date | Thanks to... |\r\n|-------------|-----------------------------------------------------------------------------------------|\r\n| 2018 | Thanks to Hepesu for help with Python3 support and ideas about breaking up calculations |\r\n| Dec 2019 | Thanks to Gareth Nisbet for allowing me to inlude his multiple scattering siumulation |\r\n| April 2020 | Thanks to ChunHai Wang for helpful suggestions in readcif! |\r\n| May 2020 | Thanks to AndreEbel for helpful suggestions on citations |\r\n| Dec 2020 | Thanks to Chris Drozdowski for suggestions about reflection families |\r\n| Jan 2021 | Thanks to aslarsen for suggestions about outputting the structure factor |\r\n| April 2021 | Thanks to Trygve R\u00e6der for suggestions about x-ray scattering factors |\r\n| Feb 2022 | Thanks to Mirko for pointing out the error in two-theta values in Scatter.powder |\r\n| March 2022 | Thanks to yevgenyr for suggesting new peak profiles in Scatter.powder |\r\n| Jan 2023 | Thanks to Anuradha Vibhakar for pointing out the error in f0 + if'-if'' |\r\n| Jan 2023 | Thanks to Andreas Rosnes for testing the installation in jupyterlab |\r\n| May 2023 | Thanks to Carmelo Prestipino for adding electron scattering factors |\r\n| June 2023 | Thanks to Sergio I. Rincon for pointing out the rounding error in Scatter.powder |\r\n| July 2023 | Thanks to asteppke for suggested update to Arrow3D for matplotlib V>3.4 |\r\n| July 2023 | Thanks to Yves Joly for helpful suggestions on FDMNES wrapper |\r\n| Jan 2024 | Thanks to Carmelo Prestipino for adding search_distance and plot_distance | \r\n| April 2024 | Thanks to Innbig for pointing out an issue with liquid crystal simulations |\r\n| May 2024 | Thanks to paul-cares pointing out a silly spelling error in the title! |\r\n\r\nCopyright\r\n-----------------------------------------------------------------------------\r\n Copyright 2024 Diamond Light Source Ltd.\r\n\r\n Licensed under the Apache License, Version 2.0 (the \"License\");\r\n you may not use this file except in compliance with the License.\r\n You may obtain a copy of the License at\r\n\r\n http://www.apache.org/licenses/LICENSE-2.0\r\n\r\n Unless required by applicable law or agreed to in writing, software\r\n distributed under the License is distributed on an \"AS IS\" BASIS,\r\n WITHOUT WARRANTIES OR CONDITIONS OF ANY KIND, either express or implied.\r\n See the License for the specific language governing permissions and\r\n limitations under the License.\r\n\r\nFiles in this package covered by this licence:\r\n* classes_crystal.py\r\n* classes_scattering.py\r\n* classes_plotting.py\r\n* classes_properties.py\r\n* classes_multicrystal.py\r\n* classes_orientation.py\r\n* classes_orbitals.py\r\n* functions_general.py\r\n* functions_plotting.py\r\n* functions_scattering.py\r\n* functions_crystallography.py\r\n* tkgui/*.py\r\n\r\nOther files are either covered by their own licence or not licenced for other use.\r\n\r\n| Dr Daniel G Porter | [dan.porter@diamond.ac.uk](mailto:dan.porter@diamond.ac.uk) |\r\n| ---- | ---- |\r\n| [www.diamond.ac.uk](www.diamond.ac.uk) | Diamond Light Source, Chilton, Didcot, Oxon, OX11 0DE, U.K. |\r\n",
"bugtrack_url": null,
"license": "Apache License Version 2.0, January 2004 http://www.apache.org/licenses/ TERMS AND CONDITIONS FOR USE, REPRODUCTION, AND DISTRIBUTION 1. Definitions. \"License\" shall mean the terms and conditions for use, reproduction, and distribution as defined by Sections 1 through 9 of this document. \"Licensor\" shall mean the copyright owner or entity authorized by the copyright owner that is granting the License. \"Legal Entity\" shall mean the union of the acting entity and all other entities that control, are controlled by, or are under common control with that entity. For the purposes of this definition, \"control\" means (i) the power, direct or indirect, to cause the direction or management of such entity, whether by contract or otherwise, or (ii) ownership of fifty percent (50%) or more of the outstanding shares, or (iii) beneficial ownership of such entity. \"You\" (or \"Your\") shall mean an individual or Legal Entity exercising permissions granted by this License. \"Source\" form shall mean the preferred form for making modifications, including but not limited to software source code, documentation source, and configuration files. \"Object\" form shall mean any form resulting from mechanical transformation or translation of a Source form, including but not limited to compiled object code, generated documentation, and conversions to other media types. \"Work\" shall mean the work of authorship, whether in Source or Object form, made available under the License, as indicated by a copyright notice that is included in or attached to the work (an example is provided in the Appendix below). \"Derivative Works\" shall mean any work, whether in Source or Object form, that is based on (or derived from) the Work and for which the editorial revisions, annotations, elaborations, or other modifications represent, as a whole, an original work of authorship. For the purposes of this License, Derivative Works shall not include works that remain separable from, or merely link (or bind by name) to the interfaces of, the Work and Derivative Works thereof. \"Contribution\" shall mean any work of authorship, including the original version of the Work and any modifications or additions to that Work or Derivative Works thereof, that is intentionally submitted to Licensor for inclusion in the Work by the copyright owner or by an individual or Legal Entity authorized to submit on behalf of the copyright owner. For the purposes of this definition, \"submitted\" means any form of electronic, verbal, or written communication sent to the Licensor or its representatives, including but not limited to communication on electronic mailing lists, source code control systems, and issue tracking systems that are managed by, or on behalf of, the Licensor for the purpose of discussing and improving the Work, but excluding communication that is conspicuously marked or otherwise designated in writing by the copyright owner as \"Not a Contribution.\" \"Contributor\" shall mean Licensor and any individual or Legal Entity on behalf of whom a Contribution has been received by Licensor and subsequently incorporated within the Work. 2. Grant of Copyright License. Subject to the terms and conditions of this License, each Contributor hereby grants to You a perpetual, worldwide, non-exclusive, no-charge, royalty-free, irrevocable copyright license to reproduce, prepare Derivative Works of, publicly display, publicly perform, sublicense, and distribute the Work and such Derivative Works in Source or Object form. 3. Grant of Patent License. Subject to the terms and conditions of this License, each Contributor hereby grants to You a perpetual, worldwide, non-exclusive, no-charge, royalty-free, irrevocable (except as stated in this section) patent license to make, have made, use, offer to sell, sell, import, and otherwise transfer the Work, where such license applies only to those patent claims licensable by such Contributor that are necessarily infringed by their Contribution(s) alone or by combination of their Contribution(s) with the Work to which such Contribution(s) was submitted. If You institute patent litigation against any entity (including a cross-claim or counterclaim in a lawsuit) alleging that the Work or a Contribution incorporated within the Work constitutes direct or contributory patent infringement, then any patent licenses granted to You under this License for that Work shall terminate as of the date such litigation is filed. 4. Redistribution. You may reproduce and distribute copies of the Work or Derivative Works thereof in any medium, with or without modifications, and in Source or Object form, provided that You meet the following conditions: (a) You must give any other recipients of the Work or Derivative Works a copy of this License; and (b) You must cause any modified files to carry prominent notices stating that You changed the files; and (c) You must retain, in the Source form of any Derivative Works that You distribute, all copyright, patent, trademark, and attribution notices from the Source form of the Work, excluding those notices that do not pertain to any part of the Derivative Works; and (d) If the Work includes a \"NOTICE\" text file as part of its distribution, then any Derivative Works that You distribute must include a readable copy of the attribution notices contained within such NOTICE file, excluding those notices that do not pertain to any part of the Derivative Works, in at least one of the following places: within a NOTICE text file distributed as part of the Derivative Works; within the Source form or documentation, if provided along with the Derivative Works; or, within a display generated by the Derivative Works, if and wherever such third-party notices normally appear. The contents of the NOTICE file are for informational purposes only and do not modify the License. You may add Your own attribution notices within Derivative Works that You distribute, alongside or as an addendum to the NOTICE text from the Work, provided that such additional attribution notices cannot be construed as modifying the License. You may add Your own copyright statement to Your modifications and may provide additional or different license terms and conditions for use, reproduction, or distribution of Your modifications, or for any such Derivative Works as a whole, provided Your use, reproduction, and distribution of the Work otherwise complies with the conditions stated in this License. 5. Submission of Contributions. Unless You explicitly state otherwise, any Contribution intentionally submitted for inclusion in the Work by You to the Licensor shall be under the terms and conditions of this License, without any additional terms or conditions. Notwithstanding the above, nothing herein shall supersede or modify the terms of any separate license agreement you may have executed with Licensor regarding such Contributions. 6. Trademarks. This License does not grant permission to use the trade names, trademarks, service marks, or product names of the Licensor, except as required for reasonable and customary use in describing the origin of the Work and reproducing the content of the NOTICE file. 7. Disclaimer of Warranty. Unless required by applicable law or agreed to in writing, Licensor provides the Work (and each Contributor provides its Contributions) on an \"AS IS\" BASIS, WITHOUT WARRANTIES OR CONDITIONS OF ANY KIND, either express or implied, including, without limitation, any warranties or conditions of TITLE, NON-INFRINGEMENT, MERCHANTABILITY, or FITNESS FOR A PARTICULAR PURPOSE. You are solely responsible for determining the appropriateness of using or redistributing the Work and assume any risks associated with Your exercise of permissions under this License. 8. Limitation of Liability. In no event and under no legal theory, whether in tort (including negligence), contract, or otherwise, unless required by applicable law (such as deliberate and grossly negligent acts) or agreed to in writing, shall any Contributor be liable to You for damages, including any direct, indirect, special, incidental, or consequential damages of any character arising as a result of this License or out of the use or inability to use the Work (including but not limited to damages for loss of goodwill, work stoppage, computer failure or malfunction, or any and all other commercial damages or losses), even if such Contributor has been advised of the possibility of such damages. 9. Accepting Warranty or Additional Liability. While redistributing the Work or Derivative Works thereof, You may choose to offer, and charge a fee for, acceptance of support, warranty, indemnity, or other liability obligations and/or rights consistent with this License. However, in accepting such obligations, You may act only on Your own behalf and on Your sole responsibility, not on behalf of any other Contributor, and only if You agree to indemnify, defend, and hold each Contributor harmless for any liability incurred by, or claims asserted against, such Contributor by reason of your accepting any such warranty or additional liability. END OF TERMS AND CONDITIONS APPENDIX: How to apply the Apache License to your work. To apply the Apache License to your work, attach the following boilerplate notice, with the fields enclosed by brackets \"[]\" replaced with your own identifying information. (Don't include the brackets!) The text should be enclosed in the appropriate comment syntax for the file format. We also recommend that a file or class name and description of purpose be included on the same \"printed page\" as the copyright notice for easier identification within third-party archives. Copyright [yyyy] [name of copyright owner] Licensed under the Apache License, Version 2.0 (the \"License\"); you may not use this file except in compliance with the License. You may obtain a copy of the License at http://www.apache.org/licenses/LICENSE-2.0 Unless required by applicable law or agreed to in writing, software distributed under the License is distributed on an \"AS IS\" BASIS, WITHOUT WARRANTIES OR CONDITIONS OF ANY KIND, either express or implied. See the License for the specific language governing permissions and limitations under the License. ",
"summary": "Generate diffracted intensities from crystals",
"version": "3.3.2",
"project_urls": {
"Bug Tracker": "https://github.com/DanPorter/Dans_Diffraction/issues",
"Changelog": "https://github.com/DanPorter/Dans_Diffraction/blob/master/README.md",
"Documentation": "https://danporter.github.io/Dans_Diffraction",
"Homepage": "https://github.com/DanPorter/Dans_Diffraction",
"Repository": "https://github.com/DanPorter/Dans_Diffraction"
},
"split_keywords": [
"crystal",
" cif",
" diffraction",
" crystallography",
" science",
" x-ray",
" neutron",
" resonant",
" magnetic",
" magnetism",
" multiple scattering",
" fdmnes",
" super structure",
" spacegroup",
" space group",
" diffractometer"
],
"urls": [
{
"comment_text": "",
"digests": {
"blake2b_256": "1fea0d79d04a5de345ee9d563da92837190ff025d2913bc18305aefc0f434615",
"md5": "9b811eb751300f14d3657af7c2cf7d38",
"sha256": "df1a0e79a4a8ed00b000f6c8e8770d44888597fea00770bbb0df75b4d8e57a71"
},
"downloads": -1,
"filename": "Dans_Diffraction-3.3.2-py3-none-any.whl",
"has_sig": false,
"md5_digest": "9b811eb751300f14d3657af7c2cf7d38",
"packagetype": "bdist_wheel",
"python_version": "py3",
"requires_python": ">=3.7",
"size": 3690717,
"upload_time": "2024-11-21T19:22:59",
"upload_time_iso_8601": "2024-11-21T19:22:59.956901Z",
"url": "https://files.pythonhosted.org/packages/1f/ea/0d79d04a5de345ee9d563da92837190ff025d2913bc18305aefc0f434615/Dans_Diffraction-3.3.2-py3-none-any.whl",
"yanked": false,
"yanked_reason": null
},
{
"comment_text": "",
"digests": {
"blake2b_256": "3ac56bd0f043b1de74f939ff84a0d89f78d4a40b700769ba120c112db73015a7",
"md5": "18df74e3437e349cc07fc23f441402e0",
"sha256": "caaa8cbb4ebf6fb665620ac86135469dd01befa0a09ba73907511abe56c85f8b"
},
"downloads": -1,
"filename": "dans_diffraction-3.3.2.tar.gz",
"has_sig": false,
"md5_digest": "18df74e3437e349cc07fc23f441402e0",
"packagetype": "sdist",
"python_version": "source",
"requires_python": ">=3.7",
"size": 3629530,
"upload_time": "2024-11-21T19:23:02",
"upload_time_iso_8601": "2024-11-21T19:23:02.578166Z",
"url": "https://files.pythonhosted.org/packages/3a/c5/6bd0f043b1de74f939ff84a0d89f78d4a40b700769ba120c112db73015a7/dans_diffraction-3.3.2.tar.gz",
"yanked": false,
"yanked_reason": null
}
],
"upload_time": "2024-11-21 19:23:02",
"github": true,
"gitlab": false,
"bitbucket": false,
"codeberg": false,
"github_user": "DanPorter",
"github_project": "Dans_Diffraction",
"travis_ci": false,
"coveralls": false,
"github_actions": true,
"requirements": [],
"lcname": "dans-diffraction"
}
