[](https://github.com/theislab/scib/stargazers)
[](https://pypi.org/project/scib)
[](https://pepy.tech/project/scib)
[](https://github.com/theislab/scib/actions/workflows/test.yml)
[](https://scib.readthedocs.io/en/latest/?badge=latest)
[](https://codecov.io/gh/theislab/scib)
[](https://github.com/pre-commit/pre-commit)
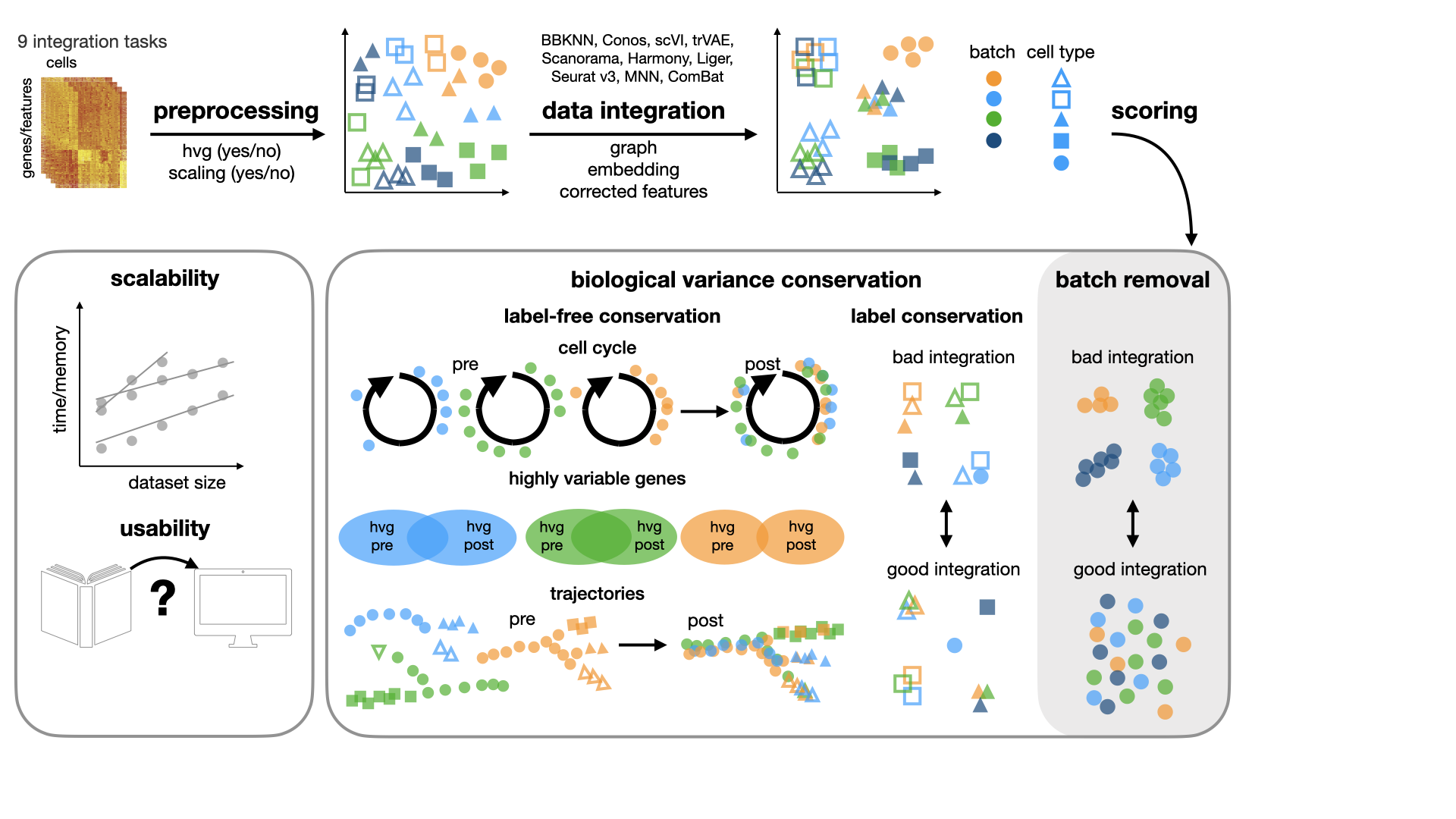
# Benchmarking atlas-level data integration in single-cell genomics
This repository contains the code for the `scib` package used in our benchmarking study for data integration tools.
In [our study](https://doi.org/10.1038/s41592-021-01336-8), we benchmark 16 methods (see Tools) with 4 combinations of
preprocessing steps leading to 68 methods combinations on 85 batches of gene expression and chromatin accessibility
data.
## Resources
- The git repository of the [`scib` package](https://github.com/theislab/scib) and
its [documentation](https://scib.readthedocs.io/).
- The reusable pipeline we used in the study can be found in the
separate [scib pipeline](https://github.com/theislab/scib-pipeline.git) repository. It is reproducible and automates
the computation of preprocesssing combinations, integration methods and benchmarking metrics.
- On our [website](https://theislab.github.io/scib-reproducibility) we visualise the results of the study.
- For reproducibility and visualisation we have a dedicated
repository: [scib-reproducibility](https://github.com/theislab/scib-reproducibility).
### Please cite:
Luecken, M.D., Büttner, M., Chaichoompu, K. et al. Benchmarking atlas-level data integration in single-cell genomics.
Nat Methods 19, 41–50 (2022). [https://doi.org/10.1038/s41592-021-01336-8](https://doi.org/10.1038/s41592-021-01336-8)
## Package: scib
We created the python package called `scib` that uses `scanpy` to streamline the integration of single-cell datasets and
evaluate the results. The package contains several modules for preprocessing an `anndata` object, running integration
methods and evaluating the resulting using a number of metrics. For preprocessing, `scib.preprocessing` (or `scib.pp`)
contains functions for normalising, scaling or batch-aware selection of highly variable genes. Functions for the
integration methods are in `scib.integration` or for short `scib.ig` and metrics are under
`scib.metrics` (or `scib.me`).
The `scib` python package is available on [PyPI](https://pypi.org/) and can be installed through
```commandline
pip install scib
```
Import `scib` in python:
```python
import scib
```
### Optional Dependencies
The package contains optional dependencies that need to be installed manually if needed.
These include R dependencies (`rpy2`, `anndata2ri`) which require an installation of R integration method packages.
All optional dependencies are listed under `setup.cfg` under `[options.extras_require]` and can be installed through pip.
e.g. for installing `rpy2` and `bbknn` dependencies:
```commandline
pip install 'scib[rpy2,bbknn]'
```
Optional dependencies outside of python need to be installed separately.
For instance, in order to run kBET, install it via the following command in R:
```R
install.packages('remotes')
remotes::install_github('theislab/kBET')
```
## Metrics
We implemented different metrics for evaluating batch correction and biological conservation in the `scib.metrics`
module.
<table class="docutils align-default">
<colgroup>
<col style="width: 50%" />
<col style="width: 50%" />
</colgroup>
<thead>
<tr class="row-odd"><th class="head"><p>Biological Conservation</p></th>
<th class="head"><p>Batch Correction</p></th>
</tr>
</thead>
<tbody>
<tr class="row-even" >
<td><ul class="simple">
<li><p>Cell type ASW</p></li>
<li><p>Cell cycle conservation</p></li>
<li><p>Graph cLISI</p></li>
<li><p>Adjusted rand index (ARI) for cell label</p></li>
<li><p>Normalised mutual information (NMI) for cell label</p></li>
<li><p>Highly variable gene conservation</p></li>
<li><p>Isolated label ASW</p></li>
<li><p>Isolated label F1</p></li>
<li><p>Trajectory conservation</p></li>
</ul></td>
<td><ul class="simple">
<li><p>Batch ASW</p></li>
<li><p>Principal component regression</p></li>
<li><p>Graph iLISI</p></li>
<li><p>Graph connectivity</p></li>
<li><p>kBET (K-nearest neighbour batch effect)</p></li>
</ul></td>
</tr>
</tbody>
</table>
For a detailed description of the metrics implemented in this package, please see our
[publication](https://doi.org/10.1038/s41592-021-01336-8) and the package [documentation](https://scib.readthedocs.io/).
## Integration Tools
Tools that are compared include:
- [BBKNN](https://github.com/Teichlab/bbknn) 1.3.9
- [Combat](https://scanpy.readthedocs.io/en/stable/api/scanpy.pp.combat.html) [paper](https://academic.oup.com/biostatistics/article/8/1/118/252073)
- [Conos](https://github.com/hms-dbmi/conos) 1.3.0
- [DESC](https://github.com/eleozzr/desc) 2.0.3
- [FastMNN](https://bioconductor.org/packages/batchelor/) (batchelor 1.4.0)
- [Harmony](https://github.com/immunogenomics/harmony) 1.0
- [LIGER](https://github.com/MacoskoLab/liger) 0.5.0
- [MNN](https://github.com/chriscainx/mnnpy) 0.1.9.5
- [SAUCIE](https://github.com/KrishnaswamyLab/SAUCIE)
- [Scanorama](https://github.com/brianhie/scanorama) 1.7.0
- [scANVI](https://github.com/chenlingantelope/HarmonizationSCANVI) (scVI 0.6.7)
- [scGen](https://github.com/theislab/scgen) 1.1.5
- [scVI](https://github.com/YosefLab/scVI) 0.6.7
- [Seurat v3](https://github.com/satijalab/seurat) 3.2.0 CCA (default) and RPCA
- [TrVae](https://github.com/theislab/trvae) 0.0.1
- [TrVaep](https://github.com/theislab/trvaep) 0.1.0
## Development
For developing this package, please make sure to install additional dependencies so that you can use `pytest` and
`pre-commit`.
```shell
pip install -e '.[test,dev]'
```
Please refer to the `setup.cfg` for more optional dependencies.
Install `pre-commit` to the repository for running it automatically every time you commit in git.
```shell
pre-commit install
```
Raw data
{
"_id": null,
"home_page": "https://github.com/theislab/scib",
"name": "scib",
"maintainer": null,
"docs_url": null,
"requires_python": ">=3.9",
"maintainer_email": null,
"keywords": "benchmarking, single cell, data integration",
"author": "Malte D. Luecken, Maren Buettner, Daniel C. Strobl, Michaela F. Mueller",
"author_email": "malte.luecken@helmholtz-muenchen.de, michaela.mueller@helmholtz-muenchen.de",
"download_url": "https://files.pythonhosted.org/packages/aa/3c/fba04785b15a74a152cd265cbdb1383bbff0bac0945f9551aab2036fceb1/scib-1.1.7.tar.gz",
"platform": null,
"description": "[](https://github.com/theislab/scib/stargazers)\n[](https://pypi.org/project/scib)\n[](https://pepy.tech/project/scib)\n[](https://github.com/theislab/scib/actions/workflows/test.yml)\n[](https://scib.readthedocs.io/en/latest/?badge=latest)\n[](https://codecov.io/gh/theislab/scib)\n[](https://github.com/pre-commit/pre-commit)\n\n# Benchmarking atlas-level data integration in single-cell genomics\n\nThis repository contains the code for the `scib` package used in our benchmarking study for data integration tools.\nIn [our study](https://doi.org/10.1038/s41592-021-01336-8), we benchmark 16 methods (see Tools) with 4 combinations of\npreprocessing steps leading to 68 methods combinations on 85 batches of gene expression and chromatin accessibility\ndata.\n\n\n\n## Resources\n\n- The git repository of the [`scib` package](https://github.com/theislab/scib) and\n its [documentation](https://scib.readthedocs.io/).\n- The reusable pipeline we used in the study can be found in the\n separate [scib pipeline](https://github.com/theislab/scib-pipeline.git) repository. It is reproducible and automates\n the computation of preprocesssing combinations, integration methods and benchmarking metrics.\n- On our [website](https://theislab.github.io/scib-reproducibility) we visualise the results of the study.\n- For reproducibility and visualisation we have a dedicated\n repository: [scib-reproducibility](https://github.com/theislab/scib-reproducibility).\n\n### Please cite:\n\nLuecken, M.D., B\u00fcttner, M., Chaichoompu, K. et al. Benchmarking atlas-level data integration in single-cell genomics.\nNat Methods 19, 41\u201350 (2022). [https://doi.org/10.1038/s41592-021-01336-8](https://doi.org/10.1038/s41592-021-01336-8)\n\n## Package: scib\n\nWe created the python package called `scib` that uses `scanpy` to streamline the integration of single-cell datasets and\nevaluate the results. The package contains several modules for preprocessing an `anndata` object, running integration\nmethods and evaluating the resulting using a number of metrics. For preprocessing, `scib.preprocessing` (or `scib.pp`)\ncontains functions for normalising, scaling or batch-aware selection of highly variable genes. Functions for the\nintegration methods are in `scib.integration` or for short `scib.ig` and metrics are under\n`scib.metrics` (or `scib.me`).\n\nThe `scib` python package is available on [PyPI](https://pypi.org/) and can be installed through\n\n```commandline\npip install scib\n```\n\nImport `scib` in python:\n\n```python\nimport scib\n```\n\n### Optional Dependencies\n\nThe package contains optional dependencies that need to be installed manually if needed.\nThese include R dependencies (`rpy2`, `anndata2ri`) which require an installation of R integration method packages.\nAll optional dependencies are listed under `setup.cfg` under `[options.extras_require]` and can be installed through pip.\n\ne.g. for installing `rpy2` and `bbknn` dependencies:\n```commandline\npip install 'scib[rpy2,bbknn]'\n```\n\nOptional dependencies outside of python need to be installed separately.\nFor instance, in order to run kBET, install it via the following command in R:\n\n```R\ninstall.packages('remotes')\nremotes::install_github('theislab/kBET')\n```\n\n## Metrics\n\nWe implemented different metrics for evaluating batch correction and biological conservation in the `scib.metrics`\nmodule.\n\n<table class=\"docutils align-default\">\n <colgroup>\n <col style=\"width: 50%\" />\n <col style=\"width: 50%\" />\n </colgroup>\n <thead>\n <tr class=\"row-odd\"><th class=\"head\"><p>Biological Conservation</p></th>\n <th class=\"head\"><p>Batch Correction</p></th>\n </tr>\n </thead>\n <tbody>\n <tr class=\"row-even\" >\n <td><ul class=\"simple\">\n <li><p>Cell type ASW</p></li>\n <li><p>Cell cycle conservation</p></li>\n <li><p>Graph cLISI</p></li>\n <li><p>Adjusted rand index (ARI) for cell label</p></li>\n <li><p>Normalised mutual information (NMI) for cell label</p></li>\n <li><p>Highly variable gene conservation</p></li>\n <li><p>Isolated label ASW</p></li>\n <li><p>Isolated label F1</p></li>\n <li><p>Trajectory conservation</p></li>\n </ul></td>\n <td><ul class=\"simple\">\n <li><p>Batch ASW</p></li>\n <li><p>Principal component regression</p></li>\n <li><p>Graph iLISI</p></li>\n <li><p>Graph connectivity</p></li>\n <li><p>kBET (K-nearest neighbour batch effect)</p></li>\n </ul></td>\n </tr>\n </tbody>\n</table>\n\nFor a detailed description of the metrics implemented in this package, please see our\n[publication](https://doi.org/10.1038/s41592-021-01336-8) and the package [documentation](https://scib.readthedocs.io/).\n\n## Integration Tools\n\nTools that are compared include:\n\n- [BBKNN](https://github.com/Teichlab/bbknn) 1.3.9\n- [Combat](https://scanpy.readthedocs.io/en/stable/api/scanpy.pp.combat.html) [paper](https://academic.oup.com/biostatistics/article/8/1/118/252073)\n- [Conos](https://github.com/hms-dbmi/conos) 1.3.0\n- [DESC](https://github.com/eleozzr/desc) 2.0.3\n- [FastMNN](https://bioconductor.org/packages/batchelor/) (batchelor 1.4.0)\n- [Harmony](https://github.com/immunogenomics/harmony) 1.0\n- [LIGER](https://github.com/MacoskoLab/liger) 0.5.0\n- [MNN](https://github.com/chriscainx/mnnpy) 0.1.9.5\n- [SAUCIE](https://github.com/KrishnaswamyLab/SAUCIE)\n- [Scanorama](https://github.com/brianhie/scanorama) 1.7.0\n- [scANVI](https://github.com/chenlingantelope/HarmonizationSCANVI) (scVI 0.6.7)\n- [scGen](https://github.com/theislab/scgen) 1.1.5\n- [scVI](https://github.com/YosefLab/scVI) 0.6.7\n- [Seurat v3](https://github.com/satijalab/seurat) 3.2.0 CCA (default) and RPCA\n- [TrVae](https://github.com/theislab/trvae) 0.0.1\n- [TrVaep](https://github.com/theislab/trvaep) 0.1.0\n\n## Development\n\nFor developing this package, please make sure to install additional dependencies so that you can use `pytest` and\n`pre-commit`.\n\n```shell\npip install -e '.[test,dev]'\n```\n\nPlease refer to the `setup.cfg` for more optional dependencies.\n\nInstall `pre-commit` to the repository for running it automatically every time you commit in git.\n\n```shell\npre-commit install\n```\n",
"bugtrack_url": null,
"license": "MIT",
"summary": "Evaluating single-cell data integration methods",
"version": "1.1.7",
"project_urls": {
"Bug Tracker": "https://github.com/theislab/scib/issues",
"Homepage": "https://github.com/theislab/scib",
"Pipeline": "https://github.com/theislab/scib-pipeline",
"Reproducibility": "https://theislab.github.io/scib-reproducibility"
},
"split_keywords": [
"benchmarking",
" single cell",
" data integration"
],
"urls": [
{
"comment_text": "",
"digests": {
"blake2b_256": "e56b92f74b30e716d5a3e5318a3c0872bb0a0fdd253f5deee6151126275d0923",
"md5": "2a19681508bef104bee20bded79470d8",
"sha256": "096fb2181471182e1ef6728b675884e7fb0611ba71330068386578dcd4ce2cc7"
},
"downloads": -1,
"filename": "scib-1.1.7-1-py3-none-any.whl",
"has_sig": false,
"md5_digest": "2a19681508bef104bee20bded79470d8",
"packagetype": "bdist_wheel",
"python_version": "py3",
"requires_python": ">=3.9",
"size": 84629,
"upload_time": "2025-01-13T18:53:24",
"upload_time_iso_8601": "2025-01-13T18:53:24.482834Z",
"url": "https://files.pythonhosted.org/packages/e5/6b/92f74b30e716d5a3e5318a3c0872bb0a0fdd253f5deee6151126275d0923/scib-1.1.7-1-py3-none-any.whl",
"yanked": false,
"yanked_reason": null
},
{
"comment_text": "",
"digests": {
"blake2b_256": "aa3cfba04785b15a74a152cd265cbdb1383bbff0bac0945f9551aab2036fceb1",
"md5": "6db1e49d9586f0c08ff1cd8899d91915",
"sha256": "3bd5fed6b89adf265c317bba1a73e9418aa94574b08fab46356f5ceb98990202"
},
"downloads": -1,
"filename": "scib-1.1.7.tar.gz",
"has_sig": false,
"md5_digest": "6db1e49d9586f0c08ff1cd8899d91915",
"packagetype": "sdist",
"python_version": "source",
"requires_python": ">=3.9",
"size": 78796,
"upload_time": "2025-01-13T18:53:25",
"upload_time_iso_8601": "2025-01-13T18:53:25.983294Z",
"url": "https://files.pythonhosted.org/packages/aa/3c/fba04785b15a74a152cd265cbdb1383bbff0bac0945f9551aab2036fceb1/scib-1.1.7.tar.gz",
"yanked": false,
"yanked_reason": null
}
],
"upload_time": "2025-01-13 18:53:25",
"github": true,
"gitlab": false,
"bitbucket": false,
"codeberg": false,
"github_user": "theislab",
"github_project": "scib",
"travis_ci": false,
"coveralls": false,
"github_actions": true,
"lcname": "scib"
}

