[](https://ci.appveyor.com/project/AlessioMilanese/motus-v2/branch/master)
[](http://bioconda.github.io/recipes/motus/README.html)
[](https://github.com/motu-tool/mOTUs_v2/blob/master/LICENSE)
[](https://anaconda.org/bioconda/motus)
mOTU profiler
========
The mOTU profiler is a computational tool that estimates relative taxonomic abundance of known and currently unknown microbial community members using metagenomic shotgun sequencing data.
Check the [wiki](https://github.com/motu-tool/mOTUs/wiki) for more information.
If you are using mOTUs, please cite:
> **Reference genome-independent taxonomic profiling of microbiomes with mOTUs3**
>
> Hans-Joachim Ruscheweyh*, Alessio Milanese*, Lucas Paoli, Nicolai Karcher, Quentin Clayssen,
> Marisa Isabell Metzger, Jakob Wirbel, Peer Bork, Daniel R. Mende, Georg Zeller# & Shinichi Sunagawa#
>
> _Microbiome_ (2022)
>
> doi: [10.1186/s40168-022-01410-z](https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-022-01410-z)
Pre-requisites
--------------
The mOTU profiler requires:
* Python 3 (or higher)
* the Burrow-Wheeler Aligner v0.7.15 or higher ([bwa](https://github.com/lh3/bwa))
* SAMtools v1.5 or higher ([link](http://www.htslib.org/download/))
In order to use the command ```snv_call``` you need:
* [metaSNV v1.0.3](https://git.embl.de/costea/metaSNV), available also on [bioconda](https://anaconda.org/bioconda/metasnv) (we assume metaSNV.py to be in the system path)
Check [installation wiki](https://github.com/motu-tool/mOTUs/wiki/Installation) to see how to install the dependencies with conda.
Installation
--------------
mOTUs can be installed either by using `pip` or via `conda`.
Installation with `conda` has the advantage that it will also download and install dependencies:
```bash
# Install in the base environment
conda install motus
# OR, create a new environment
conda create -n motu-env motus
conda activate motu-env
```
Installation with `pip`:
```bash
# Download and install mOTUs
pip install motu-profiler
# Download the mOTUs database
motus downloadDB
```
You can test that motus is intalled correctly with:
```
motus profile --test
```
Basic examples
--------------
Here is a simple example on how to obtain a taxonomic profiling from a raw read file:
```bash
motus profile -s metagenomic_sample.fastq > taxonomy_profile.txt
```
You can separate the previous call as:
```bash
motus map_tax -s metagenomic_sample.fastq -o mapped_reads.sam
motus calc_mgc -i mapped_reads.sam -o mgc_ab_table.count
motus calc_motu -i mgc_ab_table.count > taxonomy_profile.txt
rm mapped_reads.sam mgc_ab_table.count
```
The use of multiple threads (`-t`) is recommended, since bwa will finish faster. Here is an example with Paired-End reads:
```bash
motus profile -f for_sample.fastq -r rev_sample.fastq -s no_pair.fastq -t 6 > taxonomy_profile.txt
```
You can merge taxonomy files from different samples with `mOTU merge`:
```shell
motus profile -s metagenomic_sample_1.fastq -o taxonomy_profile_1.txt
motus profile -s metagenomic_sample_2.fastq -o taxonomy_profile_2.txt
motus merge -i taxonomy_profile_1.txt,taxonomy_profile_2.txt > all_sample_profiles.txt
```
You can profile samples that have been sequenced through different runs:
```shell
motus profile -f sample1_run1_for.fastq,sample1_run2_for.fastq -r sample1_run1_rev.fastq,sample1_run2_rev.fastq -s sample1_run1_single.fastq > taxonomy_profile.txt
```
How mOTUs works
--------------
The mOTUs tool performs taxonomic profiling of metagenomics and metatrancriptomics samples, i.e. it identifies species and their relative abundance present in a sample. It is based on a set of mOTUs (~species) contained in the mOTUs database.
The mOTUs database is created from reference genomes, metagenomic samples and metagenome assembled genomes (MAGs):
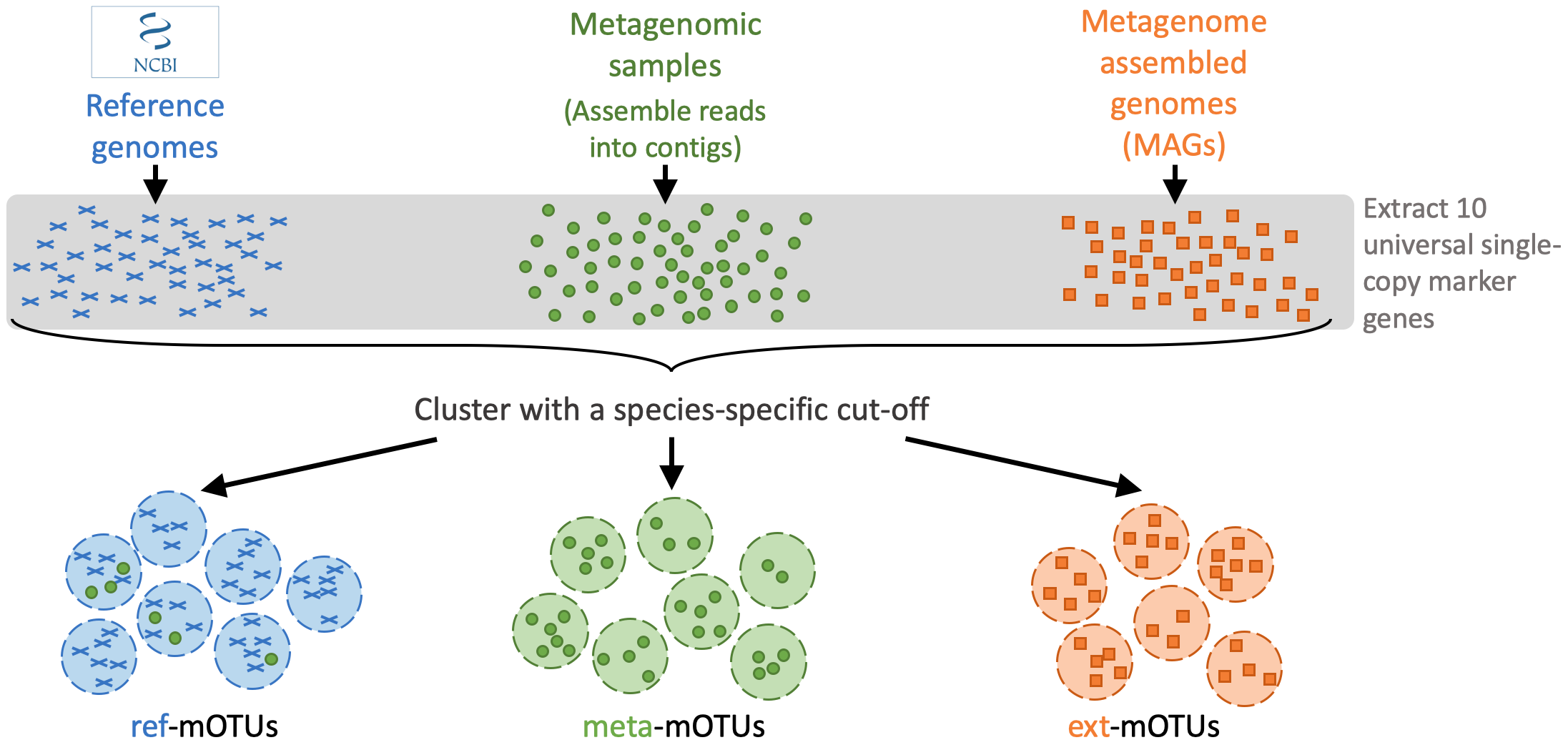
A mOTUs database is composed of three types of mOTUs:
- ref-mOTUs, which represent **known species**,
- meta-mOTUs, which represent **unknown species** obtained from metagenomic samples,
- ext-mOTUs, which represent **unknown species** obtained from MAGs.
Note that meta- and ext-mOTUs will not have a species level annotation.
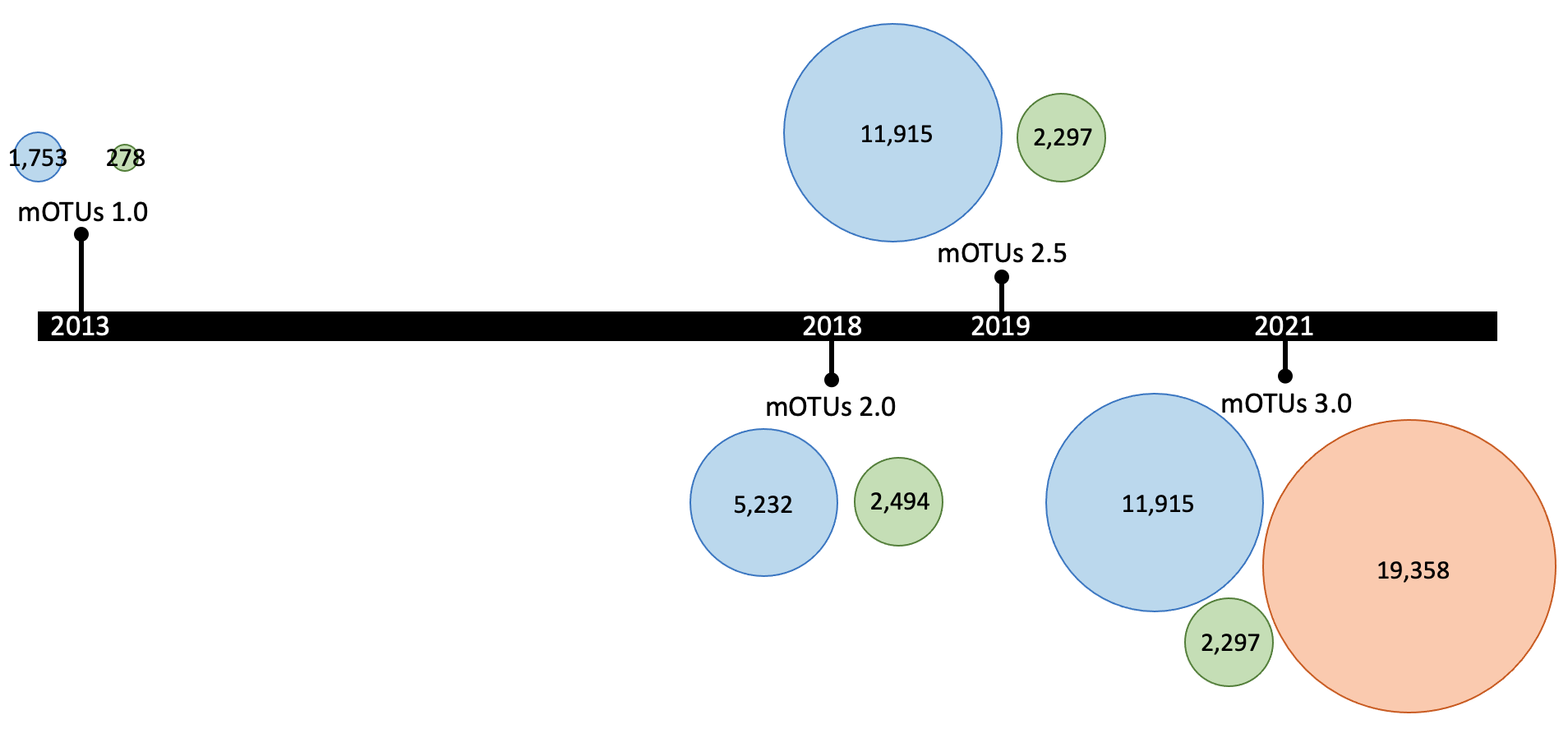
The mOTUs database is updated periodically, e.g the latest version (3.0.3), which doubles the number of profilable species by including ~600,000 draft genomes. Major releases are represented in the following graph (where the numbers represents the number of mOTUs for each of the three groups, with the same color-code as the previous graph):
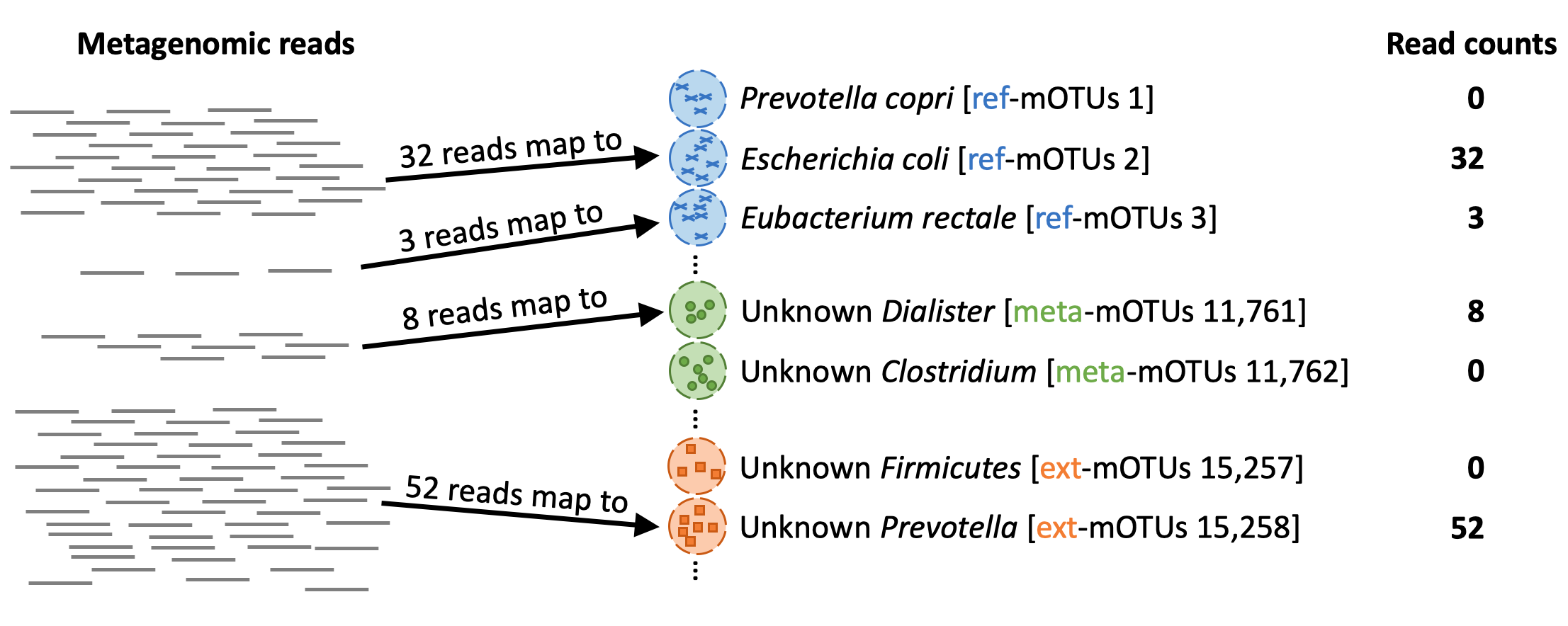
When profiling (`motus profile`) a metagenomic sample, the mOTUs tool maps the reads from the sample to the genes in the different mOTUs:
ChangeLog
--------------
**Version 3.1.0 2023-03-28 by AlessioMilanese**
* Improve database clustering algorithm and update the database (change the number of ext-mOTUs from 19,358 to 20,128)
**Version 3.0.3 2022-07-13 by AlessioMilanese**
* Add command `prep_long` to allow the profiling of long reads (more information [here](https://github.com/motu-tool/mOTUs/wiki/Profile-long-reads))
**Version 3.0.2 2022-01-31 by AlessioMilanese**
* Convert the repository to a python package and submit to PyPI
**Version 3.0.1 2021-07-27 by AlessioMilanese**
* Improve ref-mOTUs taxonomy according to #76
* Solve bug with `-A` option
**Version 3.0.0 2021-06-22 by AlessioMilanese**
* Improve code base
* Minor bug fixes
**Version 2.6.1 2021-04-27 by AlessioMilanese**
* Minor bug fixes
* Improved the taxonomy of 32 ref-mOTUs (#45)
**Version 2.6.0 2021-03-08 by AlessioMilanese**
* Add 19,358 new mOTUs
* Add taxonomic profiles of > 11k metagenomic and metatranscriptomic samples. The updated merge function can integrate those in to the users results.
* Minor bug fixes
* Change `-1` to `unassigned`
**Version 2.5.1 2019-08-17 by AlessioMilanese**
* Update the taxonomy to participate to the CAMI 2 challenge
**Version 2.5.0 2019-08-09 by AlessioMilanese**
* Add -db option to use a database from another directory
* Add -A to print all taxonomy levels together
* Update the database with more than 60k new reference genomes. There are 11,915 ref-mOTUs and 2,297 meta-mOTUs.
**Version 2.1.1 2019-03-04 by AlessioMilanese**
* Correct problem with samtools when installing with conda
**Version 2.1.0 2019-03-03 by AlessioMilanese**
* Correct error \'\t\t\' when printing -C recall
* Update database (gene coordinates)
**Version 2.0.1 2018-08-23 by AlessioMilanese**
* Add -C to print the result in CAMI format (BioBoxes format 0.9.1)
* Add -K to snv_call command to keep all the directories produced by metaSNV
**Version 2.0.0 2018-06-12 by AlessioMilanese**
* Set relative abundances as default (instead of counts)
* Add -B to print the result in BIOM format
* Add test directory
* Python2 is not supported anymore
* Minor bug fixes
**Version 2.0.0-rc1 2018-05-10 by AlessioMilanese**
* First release supporting all basic functionality.
Raw data
{
"_id": null,
"home_page": "https://github.com/motu-tool/mOTUs",
"name": "motu-profiler",
"maintainer": "",
"docs_url": null,
"requires_python": "",
"maintainer_email": "",
"keywords": "bioinformatics metagenomics taxonomic profiling",
"author": "Alessio Milanese",
"author_email": "alessiom@ethz.ch",
"download_url": "https://files.pythonhosted.org/packages/82/22/45a94f7adb2c226f013c4c00e881c3d6a9b3f7badf3f3c1d36fc8570fa1d/motu-profiler-3.1.0.tar.gz",
"platform": null,
"description": "\n\n[](https://ci.appveyor.com/project/AlessioMilanese/motus-v2/branch/master)\n[](http://bioconda.github.io/recipes/motus/README.html)\n[](https://github.com/motu-tool/mOTUs_v2/blob/master/LICENSE)\n[](https://anaconda.org/bioconda/motus)\n\n\nmOTU profiler\n========\n\nThe mOTU profiler is a computational tool that estimates relative taxonomic abundance of known and currently unknown microbial community members using metagenomic shotgun sequencing data.\n\nCheck the [wiki](https://github.com/motu-tool/mOTUs/wiki) for more information.\n\nIf you are using mOTUs, please cite:\n\n> **Reference genome-independent taxonomic profiling of microbiomes with mOTUs3**\n> \n> Hans-Joachim Ruscheweyh*, Alessio Milanese*, Lucas Paoli, Nicolai Karcher, Quentin Clayssen,\n> Marisa Isabell Metzger, Jakob Wirbel, Peer Bork, Daniel R. Mende, Georg Zeller# & Shinichi Sunagawa#\n> \n> _Microbiome_ (2022)\n> \n> doi: [10.1186/s40168-022-01410-z](https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-022-01410-z)\n\n\n\n\nPre-requisites\n--------------\n\nThe mOTU profiler requires:\n* Python 3 (or higher)\n* the Burrow-Wheeler Aligner v0.7.15 or higher ([bwa](https://github.com/lh3/bwa))\n* SAMtools v1.5 or higher ([link](http://www.htslib.org/download/))\n\nIn order to use the command ```snv_call``` you need:\n* [metaSNV v1.0.3](https://git.embl.de/costea/metaSNV), available also on [bioconda](https://anaconda.org/bioconda/metasnv) (we assume metaSNV.py to be in the system path)\n\nCheck [installation wiki](https://github.com/motu-tool/mOTUs/wiki/Installation) to see how to install the dependencies with conda.\n\nInstallation\n--------------\n\nmOTUs can be installed either by using `pip` or via `conda`.\nInstallation with `conda` has the advantage that it will also download and install dependencies:\n```bash\n# Install in the base environment\nconda install motus\n\n# OR, create a new environment\nconda create -n motu-env motus\nconda activate motu-env\n```\n\nInstallation with `pip`:\n```bash\n# Download and install mOTUs\npip install motu-profiler\n# Download the mOTUs database\nmotus downloadDB\n```\n\nYou can test that motus is intalled correctly with:\n```\nmotus profile --test\n```\n\nBasic examples\n--------------\nHere is a simple example on how to obtain a taxonomic profiling from a raw read file:\n\n```bash\nmotus profile -s metagenomic_sample.fastq > taxonomy_profile.txt\n```\n\nYou can separate the previous call as:\n```bash\nmotus map_tax -s metagenomic_sample.fastq -o mapped_reads.sam\nmotus calc_mgc -i mapped_reads.sam -o mgc_ab_table.count\nmotus calc_motu -i mgc_ab_table.count > taxonomy_profile.txt\nrm mapped_reads.sam mgc_ab_table.count\n```\n\n\nThe use of multiple threads (`-t`) is recommended, since bwa will finish faster. Here is an example with Paired-End reads:\n\n```bash\nmotus profile -f for_sample.fastq -r rev_sample.fastq -s no_pair.fastq -t 6 > taxonomy_profile.txt\n```\n\nYou can merge taxonomy files from different samples with `mOTU merge`:\n\n```shell\nmotus profile -s metagenomic_sample_1.fastq -o taxonomy_profile_1.txt\nmotus profile -s metagenomic_sample_2.fastq -o taxonomy_profile_2.txt\nmotus merge -i taxonomy_profile_1.txt,taxonomy_profile_2.txt > all_sample_profiles.txt\n```\n\nYou can profile samples that have been sequenced through different runs:\n```shell\nmotus profile -f sample1_run1_for.fastq,sample1_run2_for.fastq -r sample1_run1_rev.fastq,sample1_run2_rev.fastq -s sample1_run1_single.fastq > taxonomy_profile.txt\n```\n\nHow mOTUs works\n--------------\nThe mOTUs tool performs taxonomic profiling of metagenomics and metatrancriptomics samples, i.e. it identifies species and their relative abundance present in a sample. It is based on a set of mOTUs (~species) contained in the mOTUs database.\nThe mOTUs database is created from reference genomes, metagenomic samples and metagenome assembled genomes (MAGs):\n\n\n\nA mOTUs database is composed of three types of mOTUs:\n- ref-mOTUs, which represent **known species**,\n- meta-mOTUs, which represent **unknown species** obtained from metagenomic samples,\n- ext-mOTUs, which represent **unknown species** obtained from MAGs.\n\nNote that meta- and ext-mOTUs will not have a species level annotation.\n\nThe mOTUs database is updated periodically, e.g the latest version (3.0.3), which doubles the number of profilable species by including ~600,000 draft genomes. Major releases are represented in the following graph (where the numbers represents the number of mOTUs for each of the three groups, with the same color-code as the previous graph):\n\n\nWhen profiling (`motus profile`) a metagenomic sample, the mOTUs tool maps the reads from the sample to the genes in the different mOTUs:\n\n\nChangeLog\n--------------\n\n**Version 3.1.0 2023-03-28 by AlessioMilanese**\n* Improve database clustering algorithm and update the database (change the number of ext-mOTUs from 19,358 to 20,128)\n\n**Version 3.0.3 2022-07-13 by AlessioMilanese**\n* Add command `prep_long` to allow the profiling of long reads (more information [here](https://github.com/motu-tool/mOTUs/wiki/Profile-long-reads))\n\n**Version 3.0.2 2022-01-31 by AlessioMilanese**\n* Convert the repository to a python package and submit to PyPI\n\n**Version 3.0.1 2021-07-27 by AlessioMilanese**\n* Improve ref-mOTUs taxonomy according to #76\n* Solve bug with `-A` option\n\n**Version 3.0.0 2021-06-22 by AlessioMilanese**\n* Improve code base\n* Minor bug fixes\n\n**Version 2.6.1 2021-04-27 by AlessioMilanese**\n* Minor bug fixes\n* Improved the taxonomy of 32 ref-mOTUs (#45)\n\n**Version 2.6.0 2021-03-08 by AlessioMilanese**\n* Add 19,358 new mOTUs\n* Add taxonomic profiles of > 11k metagenomic and metatranscriptomic samples. The updated merge function can integrate those in to the users results.\n* Minor bug fixes\n* Change `-1` to `unassigned`\n\n**Version 2.5.1 2019-08-17 by AlessioMilanese**\n* Update the taxonomy to participate to the CAMI 2 challenge\n\n**Version 2.5.0 2019-08-09 by AlessioMilanese**\n* Add -db option to use a database from another directory\n* Add -A to print all taxonomy levels together\n* Update the database with more than 60k new reference genomes. There are 11,915 ref-mOTUs and 2,297 meta-mOTUs.\n\n**Version 2.1.1 2019-03-04 by AlessioMilanese**\n* Correct problem with samtools when installing with conda\n\n**Version 2.1.0 2019-03-03 by AlessioMilanese**\n* Correct error \\'\\t\\t\\' when printing -C recall\n* Update database (gene coordinates)\n\n**Version 2.0.1 2018-08-23 by AlessioMilanese**\n* Add -C to print the result in CAMI format (BioBoxes format 0.9.1)\n* Add -K to snv_call command to keep all the directories produced by metaSNV\n\n**Version 2.0.0 2018-06-12 by AlessioMilanese**\n* Set relative abundances as default (instead of counts)\n* Add -B to print the result in BIOM format\n* Add test directory\n* Python2 is not supported anymore\n* Minor bug fixes\n\n**Version 2.0.0-rc1 2018-05-10 by AlessioMilanese**\n* First release supporting all basic functionality.\n",
"bugtrack_url": null,
"license": "GPLv3",
"summary": "Taxonomic profiling of metagenomes from diverse environments with mOTUs3",
"version": "3.1.0",
"split_keywords": [
"bioinformatics",
"metagenomics",
"taxonomic",
"profiling"
],
"urls": [
{
"comment_text": "",
"digests": {
"blake2b_256": "822245a94f7adb2c226f013c4c00e881c3d6a9b3f7badf3f3c1d36fc8570fa1d",
"md5": "c27d5ca91b623d7bf8bffc7c44628ea0",
"sha256": "38959ae1b1b9892b2b47bda49a6abc2389f49802bb36d2055dcaf15080cef3f3"
},
"downloads": -1,
"filename": "motu-profiler-3.1.0.tar.gz",
"has_sig": false,
"md5_digest": "c27d5ca91b623d7bf8bffc7c44628ea0",
"packagetype": "sdist",
"python_version": "source",
"requires_python": null,
"size": 81224,
"upload_time": "2023-04-13T15:53:57",
"upload_time_iso_8601": "2023-04-13T15:53:57.002684Z",
"url": "https://files.pythonhosted.org/packages/82/22/45a94f7adb2c226f013c4c00e881c3d6a9b3f7badf3f3c1d36fc8570fa1d/motu-profiler-3.1.0.tar.gz",
"yanked": false,
"yanked_reason": null
}
],
"upload_time": "2023-04-13 15:53:57",
"github": true,
"gitlab": false,
"bitbucket": false,
"github_user": "motu-tool",
"github_project": "mOTUs",
"travis_ci": false,
"coveralls": false,
"github_actions": false,
"appveyor": true,
"lcname": "motu-profiler"
}

